 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAditi S. Krishnapriyan
Stability-Aware Training of Neural Network Interatomic Potentials with Differentiable Boltzmann Estimators
Feb 21, 2024
Neural network interatomic potentials (NNIPs) are an attractive alternative to ab-initio methods for molecular dynamics (MD) simulations. However, they can produce unstable simulations which sample unphysical states, limiting their usefulness for modeling phenomena occurring over longer timescales. To address these challenges, we present Stability-Aware Boltzmann Estimator (StABlE) Training, a multi-modal training procedure which combines conventional supervised training from quantum-mechanical energies and forces with reference system observables, to produce stable and accurate NNIPs. StABlE Training iteratively runs MD simulations to seek out unstable regions, and corrects the instabilities via supervision with a reference observable. The training procedure is enabled by the Boltzmann Estimator, which allows efficient computation of gradients required to train neural networks to system observables, and can detect both global and local instabilities. We demonstrate our methodology across organic molecules, tetrapeptides, and condensed phase systems, along with using three modern NNIP architectures. In all three cases, StABlE-trained models achieve significant improvements in simulation stability and recovery of structural and dynamic observables. In some cases, StABlE-trained models outperform conventional models trained on datasets 50 times larger. As a general framework applicable across NNIP architectures and systems, StABlE Training is a powerful tool for training stable and accurate NNIPs, particularly in the absence of large reference datasets.
Enabling Efficient Equivariant Operations in the Fourier Basis via Gaunt Tensor Products
Jan 18, 2024Developing equivariant neural networks for the E(3) group plays an important role in modeling 3D data across real-world applications. Enforcing this equivariance primarily involves the tensor products of irreducible representations (irreps). However, the computational complexity of such operations increases significantly as higher-order tensors are used. In this work, we propose a systematic approach to substantially accelerate the computation of the tensor products of irreps. We mathematically connect the commonly used Clebsch-Gordan coefficients to the Gaunt coefficients, which are integrals of products of three spherical harmonics. Through Gaunt coefficients, the tensor product of irreps becomes equivalent to the multiplication between spherical functions represented by spherical harmonics. This perspective further allows us to change the basis for the equivariant operations from spherical harmonics to a 2D Fourier basis. Consequently, the multiplication between spherical functions represented by a 2D Fourier basis can be efficiently computed via the convolution theorem and Fast Fourier Transforms. This transformation reduces the complexity of full tensor products of irreps from $\mathcal{O}(L^6)$ to $\mathcal{O}(L^3)$, where $L$ is the max degree of irreps. Leveraging this approach, we introduce the Gaunt Tensor Product, which serves as a new method to construct efficient equivariant operations across different model architectures. Our experiments on the Open Catalyst Project and 3BPA datasets demonstrate both the increased efficiency and improved performance of our approach.
Investigating the Behavior of Diffusion Models for Accelerating Electronic Structure Calculations
Nov 02, 2023We present an investigation into diffusion models for molecular generation, with the aim of better understanding how their predictions compare to the results of physics-based calculations. The investigation into these models is driven by their potential to significantly accelerate electronic structure calculations using machine learning, without requiring expensive first-principles datasets for training interatomic potentials. We find that the inference process of a popular diffusion model for de novo molecular generation is divided into an exploration phase, where the model chooses the atomic species, and a relaxation phase, where it adjusts the atomic coordinates to find a low-energy geometry. As training proceeds, we show that the model initially learns about the first-order structure of the potential energy surface, and then later learns about higher-order structure. We also find that the relaxation phase of the diffusion model can be re-purposed to sample the Boltzmann distribution over conformations and to carry out structure relaxations. For structure relaxations, the model finds geometries with ~10x lower energy than those produced by a classical force field for small organic molecules. Initializing a density functional theory (DFT) relaxation at the diffusion-produced structures yields a >2x speedup to the DFT relaxation when compared to initializing at structures relaxed with a classical force field.
Equation Discovery with Bayesian Spike-and-Slab Priors and Efficient Kernels
Oct 09, 2023Discovering governing equations from data is important to many scientific and engineering applications. Despite promising successes, existing methods are still challenged by data sparsity as well as noise issues, both of which are ubiquitous in practice. Moreover, state-of-the-art methods lack uncertainty quantification and/or are costly in training. To overcome these limitations, we propose a novel equation discovery method based on Kernel learning and BAyesian Spike-and-Slab priors (KBASS). We use kernel regression to estimate the target function, which is flexible, expressive, and more robust to data sparsity and noises. We combine it with a Bayesian spike-and-slab prior -- an ideal Bayesian sparse distribution -- for effective operator selection and uncertainty quantification. We develop an expectation propagation expectation-maximization (EP-EM) algorithm for efficient posterior inference and function estimation. To overcome the computational challenge of kernel regression, we place the function values on a mesh and induce a Kronecker product construction, and we use tensor algebra methods to enable efficient computation and optimization. We show the significant advantages of KBASS on a list of benchmark ODE and PDE discovery tasks.
CoarsenConf: Equivariant Coarsening with Aggregated Attention for Molecular Conformer Generation
Jun 26, 2023
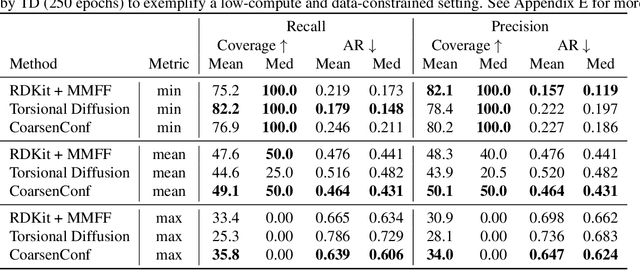
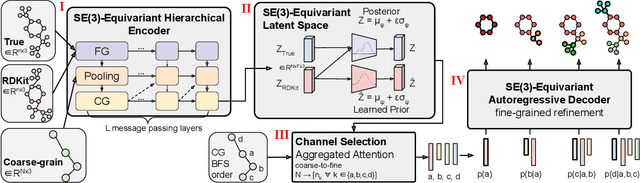
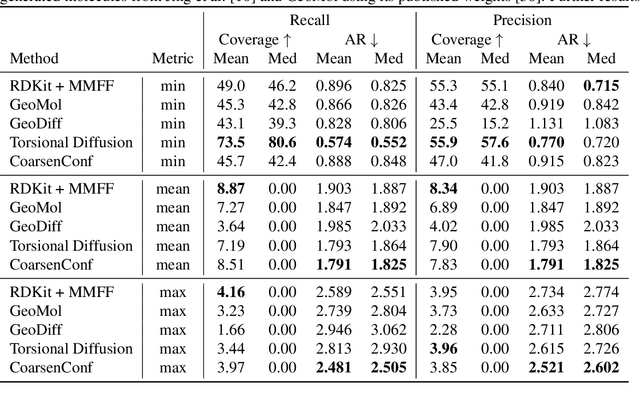
Molecular conformer generation (MCG) is an important task in cheminformatics and drug discovery. The ability to efficiently generate low-energy 3D structures can avoid expensive quantum mechanical simulations, leading to accelerated screenings and enhanced structural exploration. Several generative models have been developed for MCG, but many struggle to consistently produce high-quality conformers. To address these issues, we introduce CoarsenConf, which coarse-grains molecular graphs based on torsional angles and integrates them into an SE(3)-equivariant hierarchical variational autoencoder. Through equivariant coarse-graining, we aggregate the fine-grained atomic coordinates of subgraphs connected via rotatable bonds, creating a variable-length coarse-grained latent representation. Our model uses a novel aggregated attention mechanism to restore fine-grained coordinates from the coarse-grained latent representation, enabling efficient autoregressive generation of large molecules. Furthermore, our work expands current conformer generation benchmarks and introduces new metrics to better evaluate the quality and viability of generated conformers. We demonstrate that CoarsenConf generates more accurate conformer ensembles compared to prior generative models and traditional cheminformatics methods.
Learning differentiable solvers for systems with hard constraints
Jul 18, 2022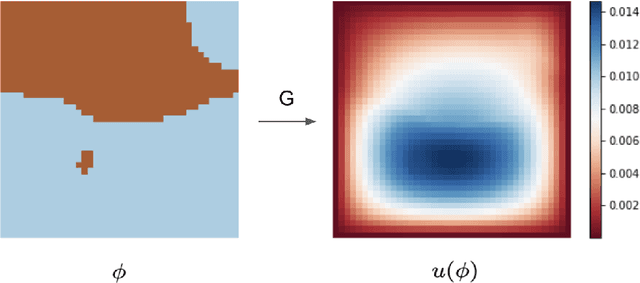
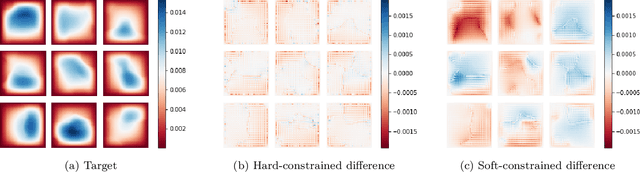
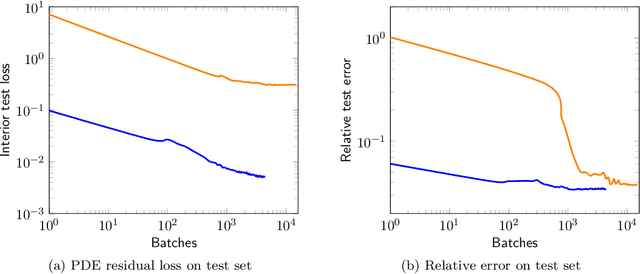
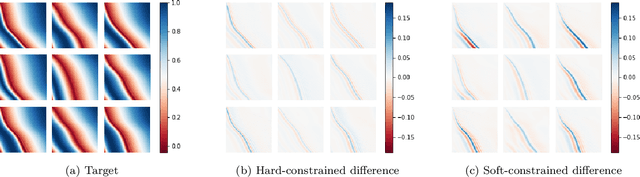
We introduce a practical method to enforce linear partial differential equation (PDE) constraints for functions defined by neural networks (NNs), up to a desired tolerance. By combining methods in differentiable physics and applications of the implicit function theorem to NN models, we develop a differentiable PDE-constrained NN layer. During training, our model learns a family of functions, each of which defines a mapping from PDE parameters to PDE solutions. At inference time, the model finds an optimal linear combination of the functions in the learned family by solving a PDE-constrained optimization problem. Our method provides continuous solutions over the domain of interest that exactly satisfy desired physical constraints. Our results show that incorporating hard constraints directly into the NN architecture achieves much lower test error, compared to training on an unconstrained objective.
Learning continuous models for continuous physics
Feb 17, 2022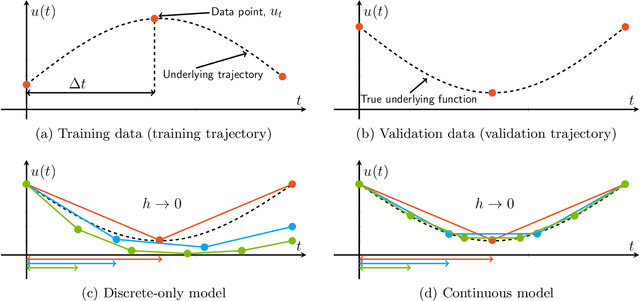

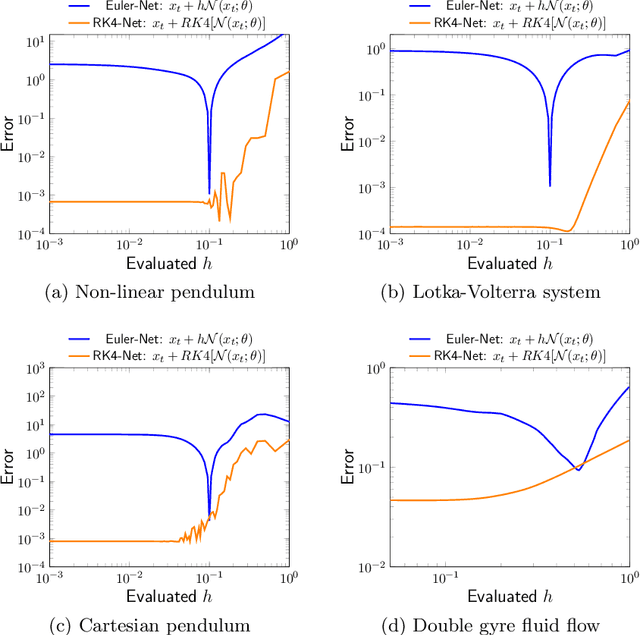
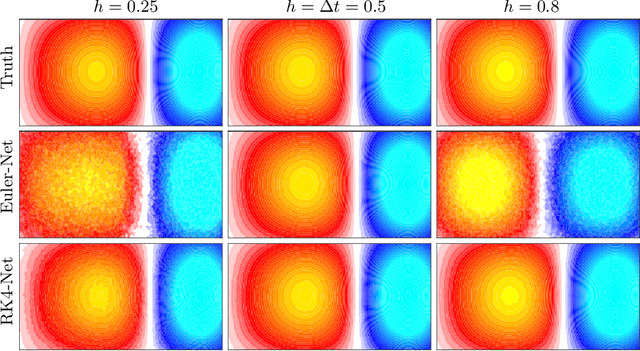
Dynamical systems that evolve continuously over time are ubiquitous throughout science and engineering. Machine learning (ML) provides data-driven approaches to model and predict the dynamics of such systems. A core issue with this approach is that ML models are typically trained on discrete data, using ML methodologies that are not aware of underlying continuity properties, which results in models that often do not capture the underlying continuous dynamics of a system of interest. As a result, these ML models are of limited use for for many scientific and engineering applications. To address this challenge, we develop a convergence test based on numerical analysis theory. Our test verifies whether a model has learned a function that accurately approximates a system's underlying continuous dynamics. Models that fail this test fail to capture relevant dynamics, rendering them of limited utility for many scientific prediction tasks; while models that pass this test enable both better interpolation and better extrapolation in multiple ways. Our results illustrate how principled numerical analysis methods can be coupled with existing ML training/testing methodologies to validate models for science and engineering applications.
Characterizing possible failure modes in physics-informed neural networks
Sep 02, 2021
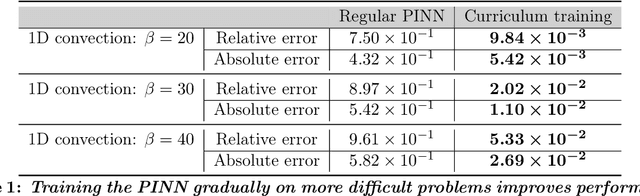

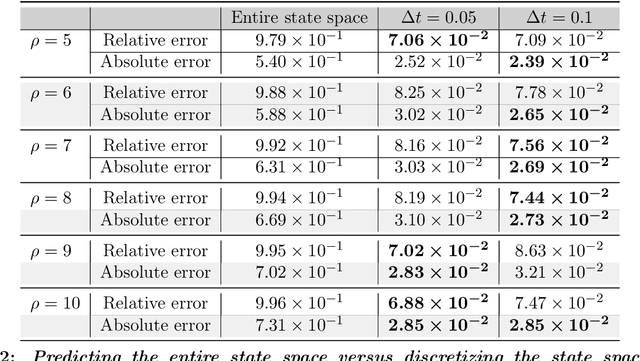
Recent work in scientific machine learning has developed so-called physics-informed neural network (PINN) models. The typical approach is to incorporate physical domain knowledge as soft constraints on an empirical loss function and use existing machine learning methodologies to train the model. We demonstrate that, while existing PINN methodologies can learn good models for relatively trivial problems, they can easily fail to learn relevant physical phenomena even for simple PDEs. In particular, we analyze several distinct situations of widespread physical interest, including learning differential equations with convection, reaction, and diffusion operators. We provide evidence that the soft regularization in PINNs, which involves differential operators, can introduce a number of subtle problems, including making the problem ill-conditioned. Importantly, we show that these possible failure modes are not due to the lack of expressivity in the NN architecture, but that the PINN's setup makes the loss landscape very hard to optimize. We then describe two promising solutions to address these failure modes. The first approach is to use curriculum regularization, where the PINN's loss term starts from a simple PDE regularization, and becomes progressively more complex as the NN gets trained. The second approach is to pose the problem as a sequence-to-sequence learning task, rather than learning to predict the entire space-time at once. Extensive testing shows that we can achieve up to 1-2 orders of magnitude lower error with these methods as compared to regular PINN training.
Topological Regularization via Persistence-Sensitive Optimization
Nov 10, 2020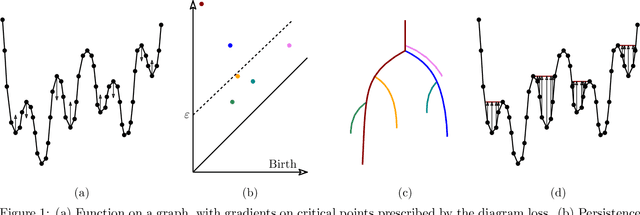

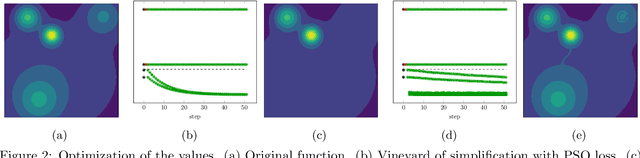

Optimization, a key tool in machine learning and statistics, relies on regularization to reduce overfitting. Traditional regularization methods control a norm of the solution to ensure its smoothness. Recently, topological methods have emerged as a way to provide a more precise and expressive control over the solution, relying on persistent homology to quantify and reduce its roughness. All such existing techniques back-propagate gradients through the persistence diagram, which is a summary of the topological features of a function. Their downside is that they provide information only at the critical points of the function. We propose a method that instead builds on persistence-sensitive simplification and translates the required changes to the persistence diagram into changes on large subsets of the domain, including both critical and regular points. This approach enables a faster and more precise topological regularization, the benefits of which we illustrate with experimental evidence.
PersGNN: Applying Topological Data Analysis and Geometric Deep Learning to Structure-Based Protein Function Prediction
Oct 30, 2020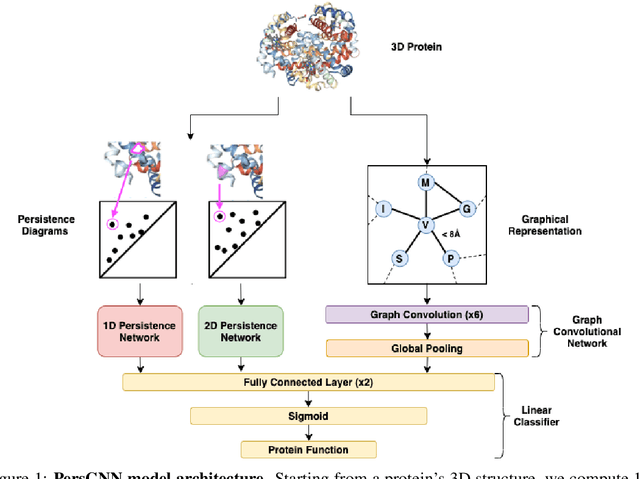

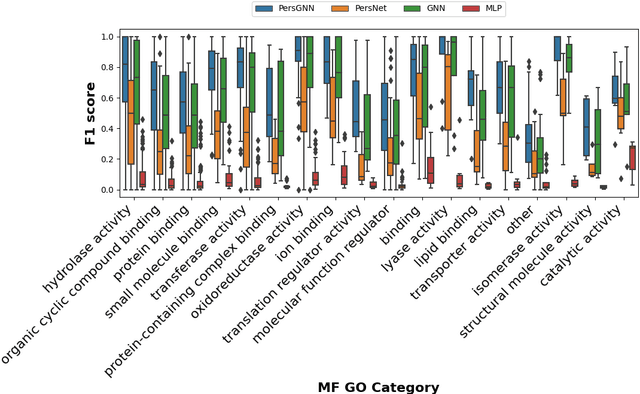

Understanding protein structure-function relationships is a key challenge in computational biology, with applications across the biotechnology and pharmaceutical industries. While it is known that protein structure directly impacts protein function, many functional prediction tasks use only protein sequence. In this work, we isolate protein structure to make functional annotations for proteins in the Protein Data Bank in order to study the expressiveness of different structure-based prediction schemes. We present PersGNN - an end-to-end trainable deep learning model that combines graph representation learning with topological data analysis to capture a complex set of both local and global structural features. While variations of these techniques have been successfully applied to proteins before, we demonstrate that our hybridized approach, PersGNN, outperforms either method on its own as well as a baseline neural network that learns from the same information. PersGNN achieves a 9.3% boost in area under the precision recall curve (AUPR) compared to the best individual model, as well as high F1 scores across different gene ontology categories, indicating the transferability of this approach.