 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEllen D. Zhong
Amortized Inference for Heterogeneous Reconstruction in Cryo-EM
Oct 13, 2022




Cryo-electron microscopy (cryo-EM) is an imaging modality that provides unique insights into the dynamics of proteins and other building blocks of life. The algorithmic challenge of jointly estimating the poses, 3D structure, and conformational heterogeneity of a biomolecule from millions of noisy and randomly oriented 2D projections in a computationally efficient manner, however, remains unsolved. Our method, cryoFIRE, performs ab initio heterogeneous reconstruction with unknown poses in an amortized framework, thereby avoiding the computationally expensive step of pose search while enabling the analysis of conformational heterogeneity. Poses and conformation are jointly estimated by an encoder while a physics-based decoder aggregates the images into an implicit neural representation of the conformational space. We show that our method can provide one order of magnitude speedup on datasets containing millions of images without any loss of accuracy. We validate that the joint estimation of poses and conformations can be amortized over the size of the dataset. For the first time, we prove that an amortized method can extract interpretable dynamic information from experimental datasets.
Explicitly disentangling image content from translation and rotation with spatial-VAE
Sep 25, 2019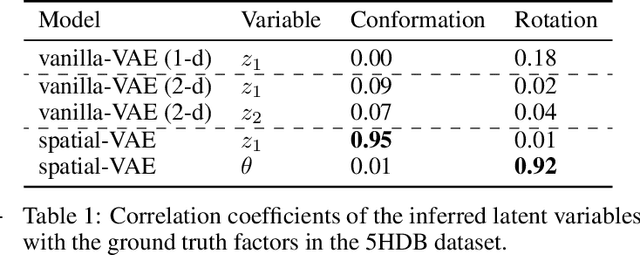
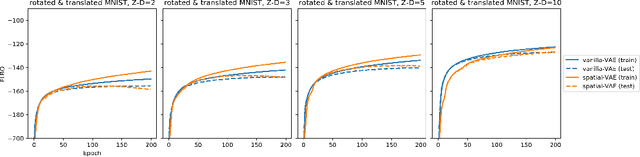
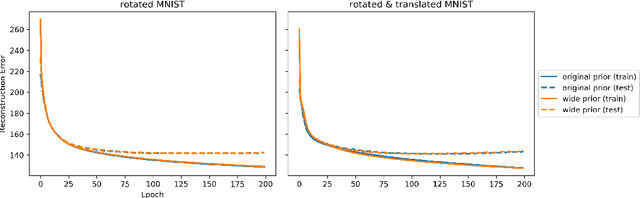
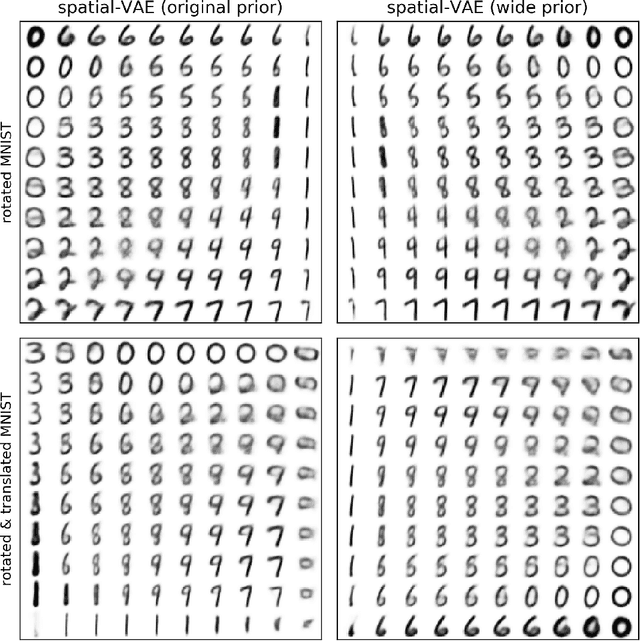
Given an image dataset, we are often interested in finding data generative factors that encode semantic content independently from pose variables such as rotation and translation. However, current disentanglement approaches do not impose any specific structure on the learned latent representations. We propose a method for explicitly disentangling image rotation and translation from other unstructured latent factors in a variational autoencoder (VAE) framework. By formulating the generative model as a function of the spatial coordinate, we make the reconstruction error differentiable with respect to latent translation and rotation parameters. This formulation allows us to train a neural network to perform approximate inference on these latent variables while explicitly constraining them to only represent rotation and translation. We demonstrate that this framework, termed spatial-VAE, effectively learns latent representations that disentangle image rotation and translation from content and improves reconstruction over standard VAEs on several benchmark datasets, including applications to modeling continuous 2-D views of proteins from single particle electron microscopy and galaxies in astronomical images.
Reconstructing continuously heterogeneous structures from single particle cryo-EM with deep generative models
Sep 11, 2019
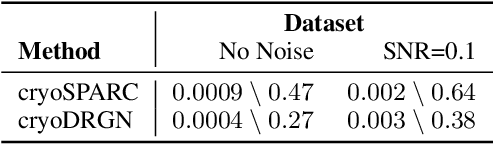
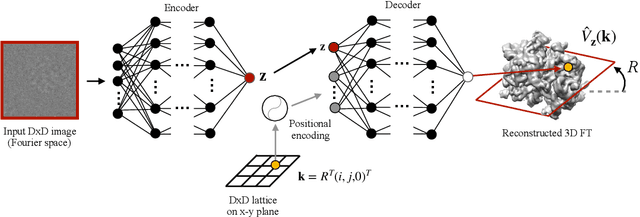
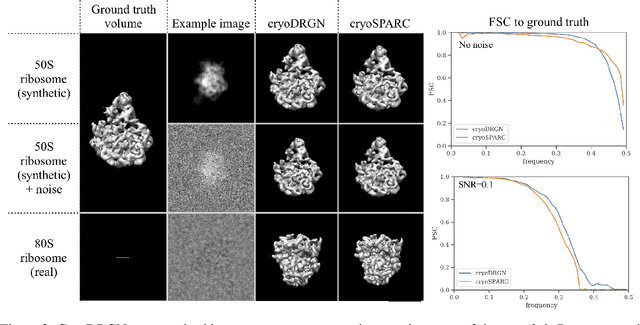
Cryo-electron microscopy (cryo-EM) is a powerful technique for determining the structure of proteins and other macromolecular complexes at near-atomic resolution. In single particle cryo-EM, the central problem is to reconstruct the three-dimensional structure of a macromolecule from $10^{4-7}$ noisy and randomly oriented two-dimensional projections. However, the imaged protein complexes may exhibit structural variability, which complicates reconstruction and is typically addressed using discrete clustering approaches that fail to capture the full range of protein dynamics. Here, we introduce a novel method for cryo-EM reconstruction that extends naturally to modeling continuous generative factors of structural heterogeneity. This method encodes structures in Fourier space using coordinate-based deep neural networks, and trains these networks from unlabeled 2D cryo-EM images by combining exact inference over image orientation with variational inference for structural heterogeneity. We demonstrate that the proposed method, termed cryoDRGN, can perform ab initio reconstruction of 3D protein complexes from simulated and real 2D cryo-EM image data. To our knowledge, cryoDRGN is the first neural network-based approach for cryo-EM reconstruction and the first end-to-end method for directly reconstructing continuous ensembles of protein structures from cryo-EM images.