 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeKen S. Lau
Nucleus subtype classification using inter-modality learning
Jan 29, 2024
Understanding the way cells communicate, co-locate, and interrelate is essential to understanding human physiology. Hematoxylin and eosin (H&E) staining is ubiquitously available both for clinical studies and research. The Colon Nucleus Identification and Classification (CoNIC) Challenge has recently innovated on robust artificial intelligence labeling of six cell types on H&E stains of the colon. However, this is a very small fraction of the number of potential cell classification types. Specifically, the CoNIC Challenge is unable to classify epithelial subtypes (progenitor, endocrine, goblet), lymphocyte subtypes (B, helper T, cytotoxic T), or connective subtypes (fibroblasts, stromal). In this paper, we propose to use inter-modality learning to label previously un-labelable cell types on virtual H&E. We leveraged multiplexed immunofluorescence (MxIF) histology imaging to identify 14 subclasses of cell types. We performed style transfer to synthesize virtual H&E from MxIF and transferred the higher density labels from MxIF to these virtual H&E images. We then evaluated the efficacy of learning in this approach. We identified helper T and progenitor nuclei with positive predictive values of $0.34 \pm 0.15$ (prevalence $0.03 \pm 0.01$) and $0.47 \pm 0.1$ (prevalence $0.07 \pm 0.02$) respectively on virtual H&E. This approach represents a promising step towards automating annotation in digital pathology.
Cell Spatial Analysis in Crohn's Disease: Unveiling Local Cell Arrangement Pattern with Graph-based Signatures
Aug 20, 2023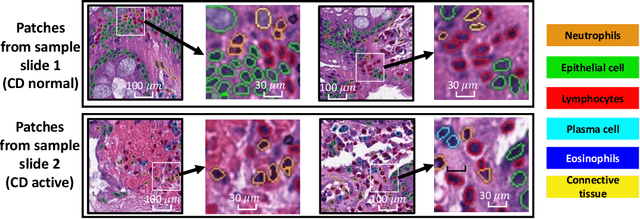
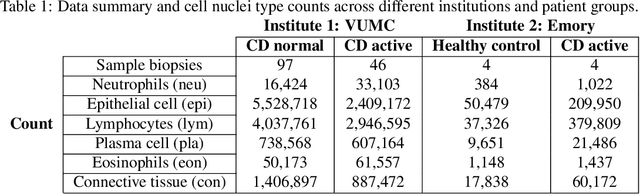
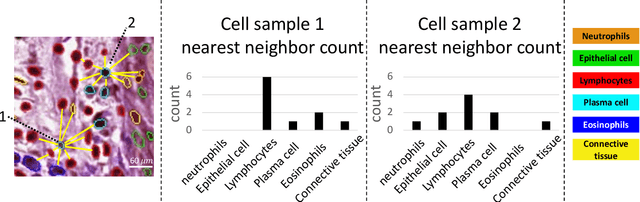
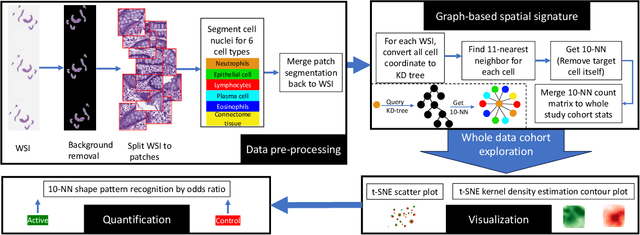
Crohn's disease (CD) is a chronic and relapsing inflammatory condition that affects segments of the gastrointestinal tract. CD activity is determined by histological findings, particularly the density of neutrophils observed on Hematoxylin and Eosin stains (H&E) imaging. However, understanding the broader morphometry and local cell arrangement beyond cell counting and tissue morphology remains challenging. To address this, we characterize six distinct cell types from H&E images and develop a novel approach for the local spatial signature of each cell. Specifically, we create a 10-cell neighborhood matrix, representing neighboring cell arrangements for each individual cell. Utilizing t-SNE for non-linear spatial projection in scatter-plot and Kernel Density Estimation contour-plot formats, our study examines patterns of differences in the cellular environment associated with the odds ratio of spatial patterns between active CD and control groups. This analysis is based on data collected at the two research institutes. The findings reveal heterogeneous nearest-neighbor patterns, signifying distinct tendencies of cell clustering, with a particular focus on the rectum region. These variations underscore the impact of data heterogeneity on cell spatial arrangements in CD patients. Moreover, the spatial distribution disparities between the two research sites highlight the significance of collaborative efforts among healthcare organizations. All research analysis pipeline tools are available at https://github.com/MASILab/cellNN.
Feasibility of Universal Anomaly Detection without Knowing the Abnormality in Medical Images
Jul 03, 2023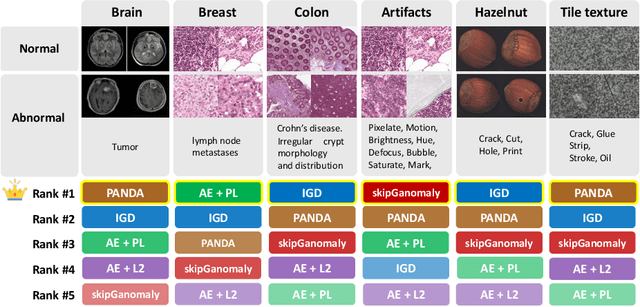
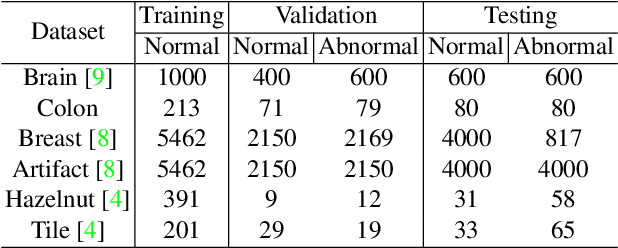
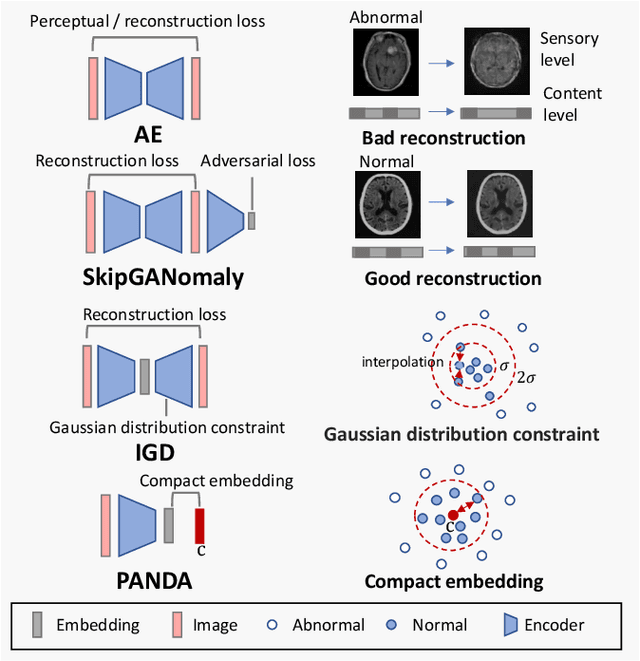
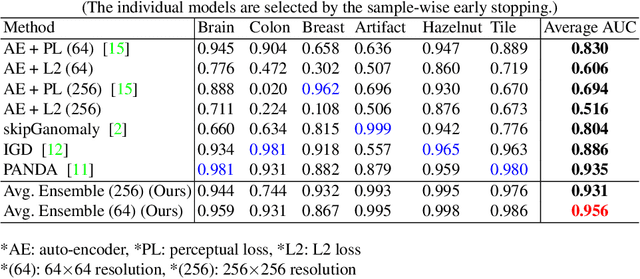
Many anomaly detection approaches, especially deep learning methods, have been recently developed to identify abnormal image morphology by only employing normal images during training. Unfortunately, many prior anomaly detection methods were optimized for a specific "known" abnormality (e.g., brain tumor, bone fraction, cell types). Moreover, even though only the normal images were used in the training process, the abnormal images were oftenly employed during the validation process (e.g., epoch selection, hyper-parameter tuning), which might leak the supposed ``unknown" abnormality unintentionally. In this study, we investigated these two essential aspects regarding universal anomaly detection in medical images by (1) comparing various anomaly detection methods across four medical datasets, (2) investigating the inevitable but often neglected issues on how to unbiasedly select the optimal anomaly detection model during the validation phase using only normal images, and (3) proposing a simple decision-level ensemble method to leverage the advantage of different kinds of anomaly detection without knowing the abnormality. The results of our experiments indicate that none of the evaluated methods consistently achieved the best performance across all datasets. Our proposed method enhanced the robustness of performance in general (average AUC 0.956).
Cross-scale Multi-instance Learning for Pathological Image Diagnosis
Apr 01, 2023

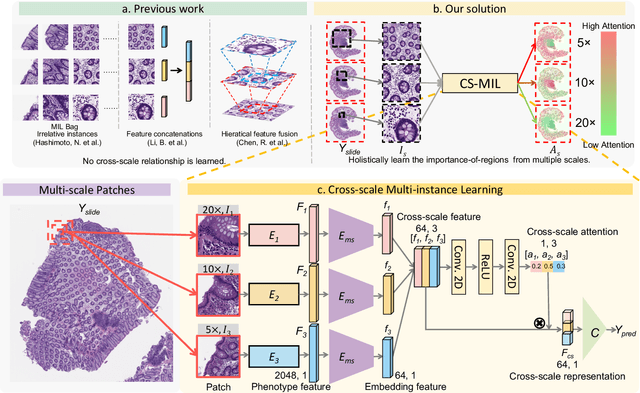

Analyzing high resolution whole slide images (WSIs) with regard to information across multiple scales poses a significant challenge in digital pathology. Multi-instance learning (MIL) is a common solution for working with high resolution images by classifying bags of objects (i.e. sets of smaller image patches). However, such processing is typically performed at a single scale (e.g., 20x magnification) of WSIs, disregarding the vital inter-scale information that is key to diagnoses by human pathologists. In this study, we propose a novel cross-scale MIL algorithm to explicitly aggregate inter-scale relationships into a single MIL network for pathological image diagnosis. The contribution of this paper is three-fold: (1) A novel cross-scale MIL (CS-MIL) algorithm that integrates the multi-scale information and the inter-scale relationships is proposed; (2) A toy dataset with scale-specific morphological features is created and released to examine and visualize differential cross-scale attention; (3) Superior performance on both in-house and public datasets is demonstrated by our simple cross-scale MIL strategy. The official implementation is publicly available at https://github.com/hrlblab/CS-MIL.
Cross-scale Attention Guided Multi-instance Learning for Crohn's Disease Diagnosis with Pathological Images
Aug 15, 2022
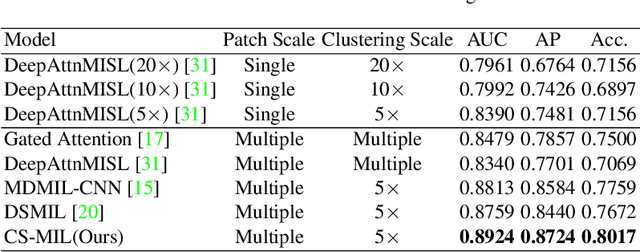
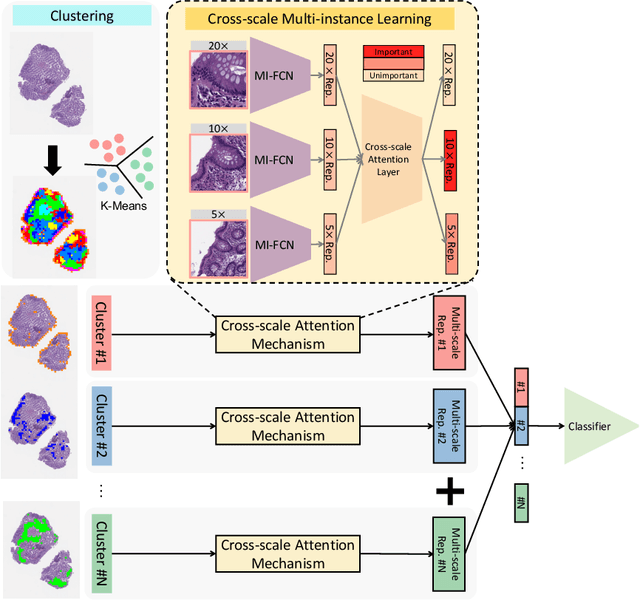
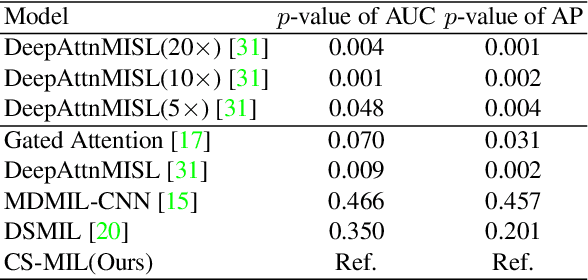
Multi-instance learning (MIL) is widely used in the computer-aided interpretation of pathological Whole Slide Images (WSIs) to solve the lack of pixel-wise or patch-wise annotations. Often, this approach directly applies "natural image driven" MIL algorithms which overlook the multi-scale (i.e. pyramidal) nature of WSIs. Off-the-shelf MIL algorithms are typically deployed on a single-scale of WSIs (e.g., 20x magnification), while human pathologists usually aggregate the global and local patterns in a multi-scale manner (e.g., by zooming in and out between different magnifications). In this study, we propose a novel cross-scale attention mechanism to explicitly aggregate inter-scale interactions into a single MIL network for Crohn's Disease (CD), which is a form of inflammatory bowel disease. The contribution of this paper is two-fold: (1) a cross-scale attention mechanism is proposed to aggregate features from different resolutions with multi-scale interaction; and (2) differential multi-scale attention visualizations are generated to localize explainable lesion patterns. By training ~250,000 H&E-stained Ascending Colon (AC) patches from 20 CD patient and 30 healthy control samples at different scales, our approach achieved a superior Area under the Curve (AUC) score of 0.8924 compared with baseline models. The official implementation is publicly available at https://github.com/hrlblab/CS-MIL.