 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMingyuan Xu
Node-based Knowledge Graph Contrastive Learning for Medical Relationship Prediction
Oct 16, 2023




The embedding of Biomedical Knowledge Graphs (BKGs) generates robust representations, valuable for a variety of artificial intelligence applications, including predicting drug combinations and reasoning disease-drug relationships. Meanwhile, contrastive learning (CL) is widely employed to enhance the distinctiveness of these representations. However, constructing suitable contrastive pairs for CL, especially within Knowledge Graphs (KGs), has been challenging. In this paper, we proposed a novel node-based contrastive learning method for knowledge graph embedding, NC-KGE. NC-KGE enhances knowledge extraction in embeddings and speeds up training convergence by constructing appropriate contrastive node pairs on KGs. This scheme can be easily integrated with other knowledge graph embedding (KGE) methods. For downstream task such as biochemical relationship prediction, we have incorporated a relation-aware attention mechanism into NC-KGE, focusing on the semantic relationships and node interactions. Extensive experiments show that NC-KGE performs competitively with state-of-the-art models on public datasets like FB15k-237 and WN18RR. Particularly in biomedical relationship prediction tasks, NC-KGE outperforms all baselines on datasets such as PharmKG8k-28, DRKG17k-21, and BioKG72k-14, especially in predicting drug combination relationships. We release our code at https://github.com/zhi520/NC-KGE.
More complex encoder is not all you need
Sep 21, 2023U-Net and its variants have been widely used in medical image segmentation. However, most current U-Net variants confine their improvement strategies to building more complex encoder, while leaving the decoder unchanged or adopting a simple symmetric structure. These approaches overlook the true functionality of the decoder: receiving low-resolution feature maps from the encoder and restoring feature map resolution and lost information through upsampling. As a result, the decoder, especially its upsampling component, plays a crucial role in enhancing segmentation outcomes. However, in 3D medical image segmentation, the commonly used transposed convolution can result in visual artifacts. This issue stems from the absence of direct relationship between adjacent pixels in the output feature map. Furthermore, plain encoder has already possessed sufficient feature extraction capability because downsampling operation leads to the gradual expansion of the receptive field, but the loss of information during downsampling process is unignorable. To address the gap in relevant research, we extend our focus beyond the encoder and introduce neU-Net (i.e., not complex encoder U-Net), which incorporates a novel Sub-pixel Convolution for upsampling to construct a powerful decoder. Additionally, we introduce multi-scale wavelet inputs module on the encoder side to provide additional information. Our model design achieves excellent results, surpassing other state-of-the-art methods on both the Synapse and ACDC datasets.
Neural Network Based in Silico Simulation of Combustion Reactions
Nov 27, 2019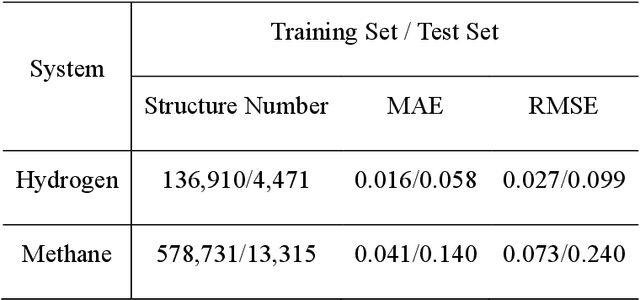
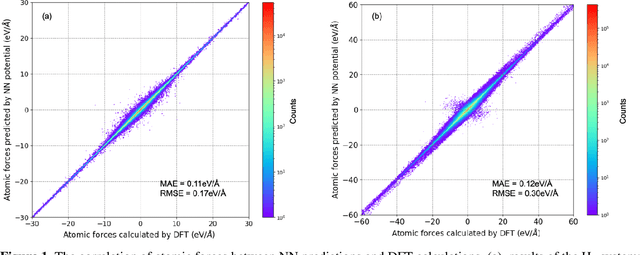
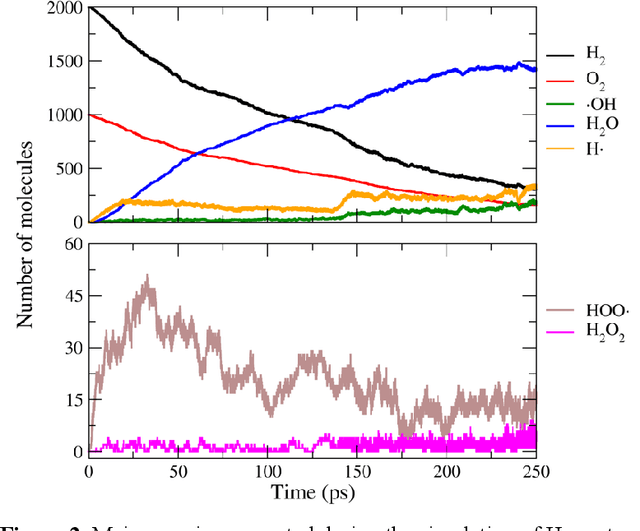
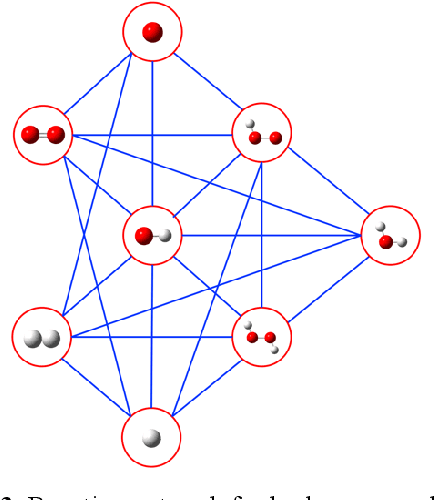
Understanding and prediction of the chemical reactions are fundamental demanding in the study of many complex chemical systems. Reactive molecular dynamics (MD) simulation has been widely used for this purpose as it can offer atomic details and can help us better interpret chemical reaction mechanisms. In this study, two reference datasets were constructed and corresponding neural network (NN) potentials were trained based on them. For given large-scale reaction systems, the NN potentials can predict the potential energy and atomic forces of DFT precision, while it is orders of magnitude faster than the conventional DFT calculation. With these two models, reactive MD simulations were performed to explore the combustion mechanisms of hydrogen and methane. Benefit from the high efficiency of the NN model, nanosecond MD trajectories for large-scale systems containing hundreds of atoms were produced and detailed combustion mechanism was obtained. Through further development, the algorithms in this study can be used to explore and discovery reaction mechanisms of many complex reaction systems, such as combustion, synthesis, and heterogeneous catalysis without any predefined reaction coordinates and elementary reaction steps.