 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMusa M. Mhlanga
Stability and Structural Properties of Gene Regulation Networks with Coregulation Rules
Apr 10, 2016
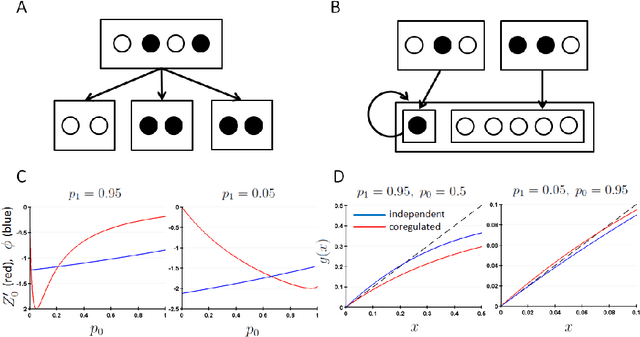
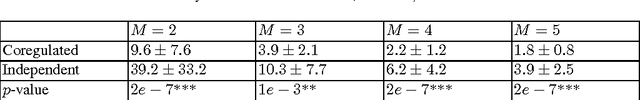
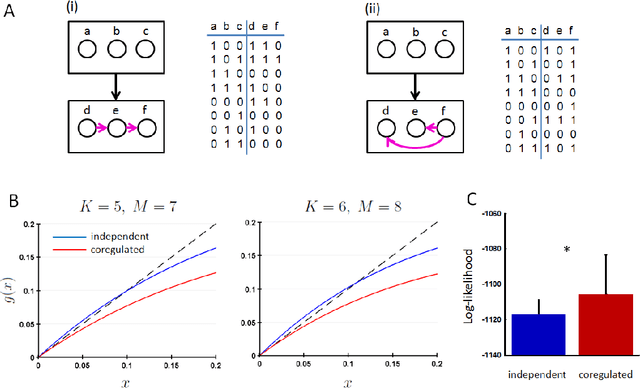
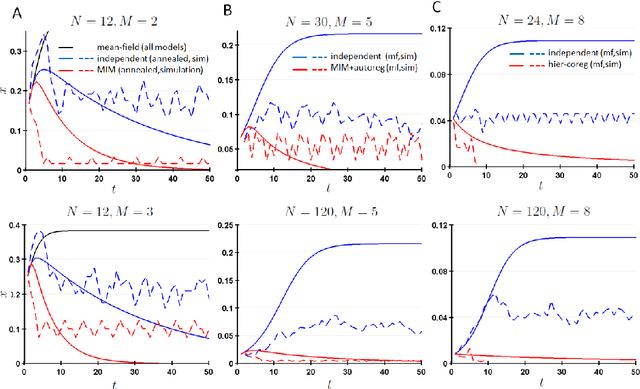
Coregulation of the expression of groups of genes has been extensively demonstrated empirically in bacterial and eukaryotic systems. Such coregulation can arise through the use of shared regulatory motifs, which allow the coordinated expression of modules (and module groups) of functionally related genes across the genome. Coregulation can also arise through the physical association of multi-gene complexes through chromosomal looping, which are then transcribed together. We present a general formalism for modeling coregulation rules in the framework of Random Boolean Networks (RBN), and develop specific models for transcription factor networks with modular structure (including module groups, and multi-input modules (MIM) with autoregulation) and multi-gene complexes (including hierarchical differentiation between multi-gene complex members). We develop a mean-field approach to analyse the stability of large networks incorporating coregulation, and show that autoregulated MIM and hierarchical gene-complex models can achieve greater stability than networks without coregulation whose rules have matching activation frequency. We provide further analysis of the stability of small networks of both kinds through simulations. We also characterize several general properties of the transients and attractors in the hierarchical coregulation model, and show using simulations that the steady-state distribution factorizes hierarchically as a Bayesian network in a Markov Jump Process analogue of the RBN model.
Generalized Statistical Tests for mRNA and Protein Subcellular Spatial Patterning against Complete Spatial Randomness
Apr 10, 2016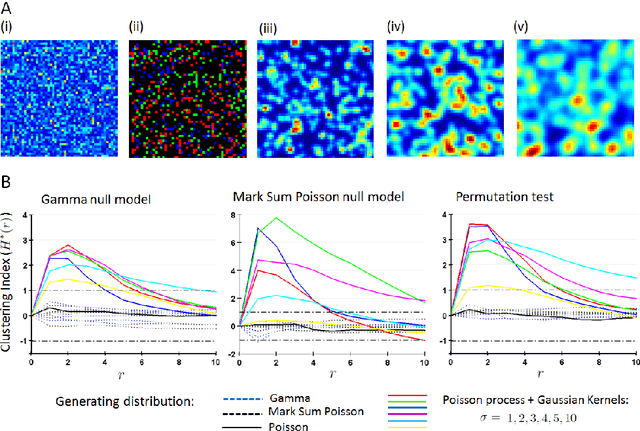
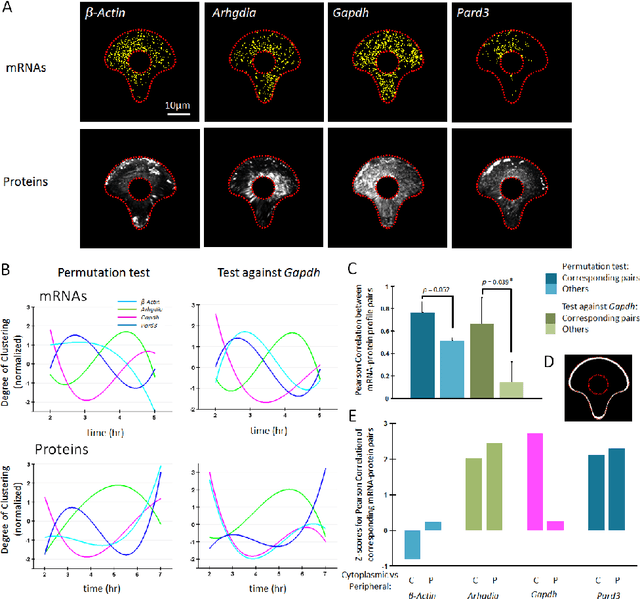
We derive generalized estimators for a number of spatial statistics that have been used in the analysis of spatially resolved omics data, such as Ripley's K, H and L functions, clustering index, and degree of clustering, which allow these statistics to be calculated on data modelled by arbitrary random measures (RMs). Our estimators generalize those typically used to calculate these statistics on point process data, allowing them to be calculated on RMs which assign continuous values to spatial regions, for instance to model protein intensity. The clustering index (H*) compares Ripley's H function calculated empirically to its distribution under complete spatial randomness (CSR), leading us to consider CSR null hypotheses for RMs which are not point-processes when generalizing this statistic. We thus consider restricted classes of completely random measures which can be simulated directly (Gamma processes and Marked Poisson Processes), as well as the general class of all CSR RMs, for which we derive an exact permutation-based H* estimator. We establish several properties of the estimators, including bounds on the accuracy of our general Ripley K estimator, its relationship to a previous estimator for the cross-correlation measure, and the relationship of our generalized H* estimator to previous statistics. To test the ability of our approach to identify spatial patterning, we use Fluorescent In Situ Hybridization (FISH) and Immunofluorescence (IF) data to probe for mRNA and protein subcellular localization patterns respectively in polarizing mouse fibroblasts on micropattened cells. We observe correlated patterns of clustering over time for corresponding mRNAs and proteins, suggesting a deterministic effect of mRNA localization on protein localization for several pairs tested, including one case in which spatial patterning at the mRNA level has not been previously demonstrated.