 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNataša Tagasovska
BOtied: Multi-objective Bayesian optimization with tied multivariate ranks
Jun 01, 2023

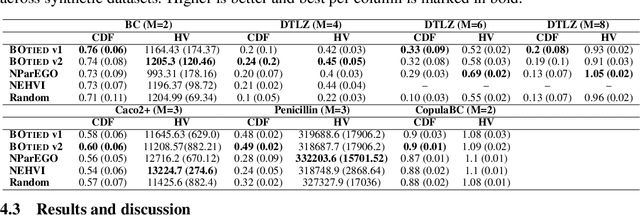
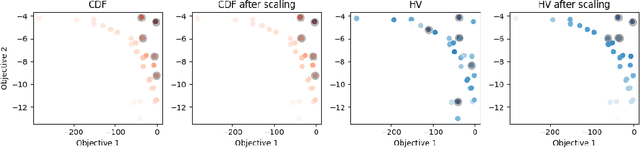

Many scientific and industrial applications require joint optimization of multiple, potentially competing objectives. Multi-objective Bayesian optimization (MOBO) is a sample-efficient framework for identifying Pareto-optimal solutions. We show a natural connection between non-dominated solutions and the highest multivariate rank, which coincides with the outermost level line of the joint cumulative distribution function (CDF). We propose the CDF indicator, a Pareto-compliant metric for evaluating the quality of approximate Pareto sets that complements the popular hypervolume indicator. At the heart of MOBO is the acquisition function, which determines the next candidate to evaluate by navigating the best compromises among the objectives. Multi-objective acquisition functions that rely on box decomposition of the objective space, such as the expected hypervolume improvement (EHVI) and entropy search, scale poorly to a large number of objectives. We propose an acquisition function, called BOtied, based on the CDF indicator. BOtied can be implemented efficiently with copulas, a statistical tool for modeling complex, high-dimensional distributions. We benchmark BOtied against common acquisition functions, including EHVI and random scalarization (ParEGO), in a series of synthetic and real-data experiments. BOtied performs on par with the baselines across datasets and metrics while being computationally efficient.
Retrospective Uncertainties for Deep Models using Vine Copulas
Feb 24, 2023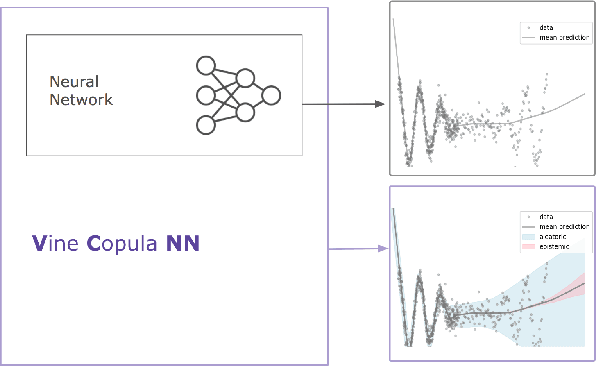
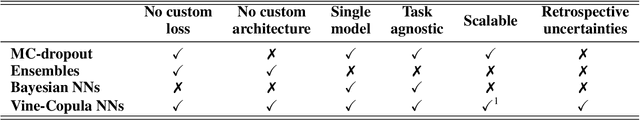
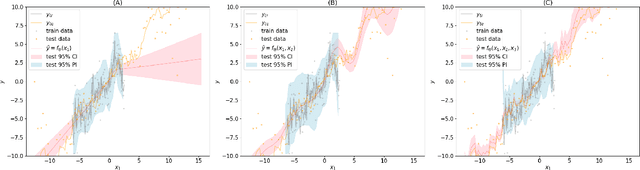

Despite the major progress of deep models as learning machines, uncertainty estimation remains a major challenge. Existing solutions rely on modified loss functions or architectural changes. We propose to compensate for the lack of built-in uncertainty estimates by supplementing any network, retrospectively, with a subsequent vine copula model, in an overall compound we call Vine-Copula Neural Network (VCNN). Through synthetic and real-data experiments, we show that VCNNs could be task (regression/classification) and architecture (recurrent, fully connected) agnostic while providing reliable and better-calibrated uncertainty estimates, comparable to state-of-the-art built-in uncertainty solutions.
Learning Causal Representations of Single Cells via Sparse Mechanism Shift Modeling
Nov 09, 2022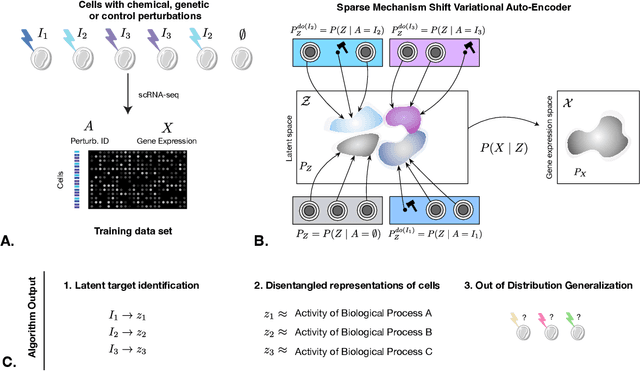
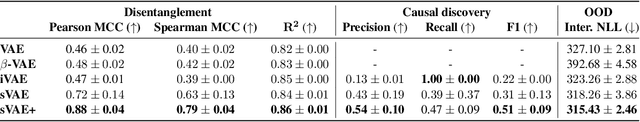
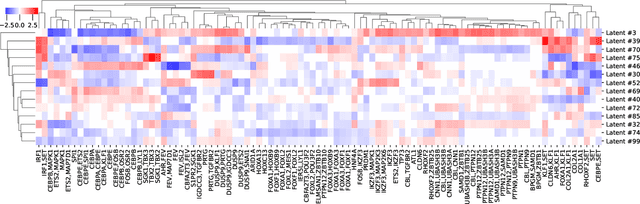
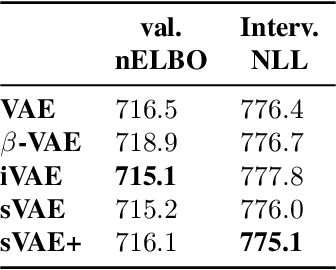
Latent variable models such as the Variational Auto-Encoder (VAE) have become a go-to tool for analyzing biological data, especially in the field of single-cell genomics. One remaining challenge is the interpretability of latent variables as biological processes that define a cell's identity. Outside of biological applications, this problem is commonly referred to as learning disentangled representations. Although several disentanglement-promoting variants of the VAE were introduced, and applied to single-cell genomics data, this task has been shown to be infeasible from independent and identically distributed measurements, without additional structure. Instead, recent methods propose to leverage non-stationary data, as well as the sparse mechanism shift assumption in order to learn disentangled representations with a causal semantic. Here, we extend the application of these methodological advances to the analysis of single-cell genomics data with genetic or chemical perturbations. More precisely, we propose a deep generative model of single-cell gene expression data for which each perturbation is treated as a stochastic intervention targeting an unknown, but sparse, subset of latent variables. We benchmark these methods on simulated single-cell data to evaluate their performance at latent units recovery, causal target identification and out-of-domain generalization. Finally, we apply those approaches to two real-world large-scale gene perturbation data sets and find that models that exploit the sparse mechanism shift hypothesis surpass contemporary methods on a transfer learning task. We implement our new model and benchmarks using the scvi-tools library, and release it as open-source software at https://github.com/Genentech/sVAE.
A Pareto-optimal compositional energy-based model for sampling and optimization of protein sequences
Oct 19, 2022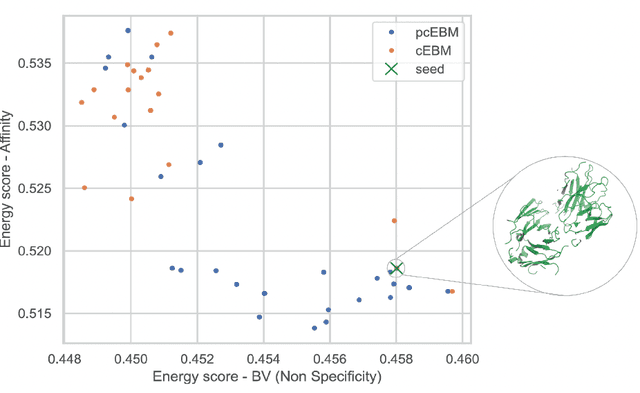
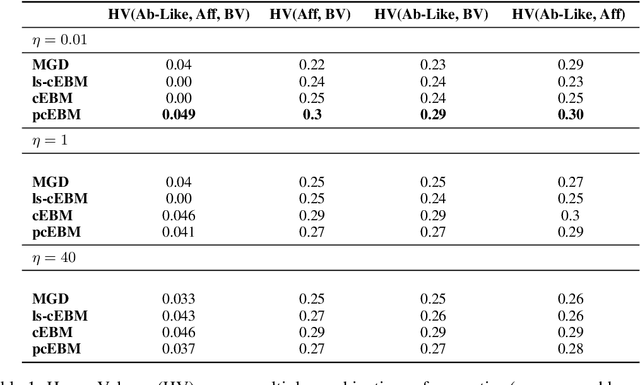
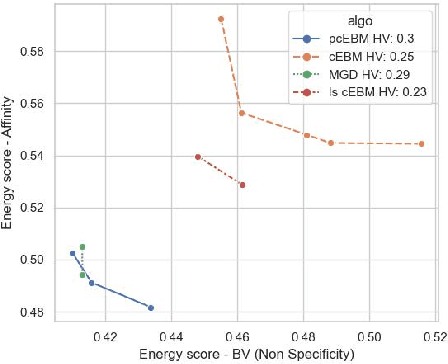
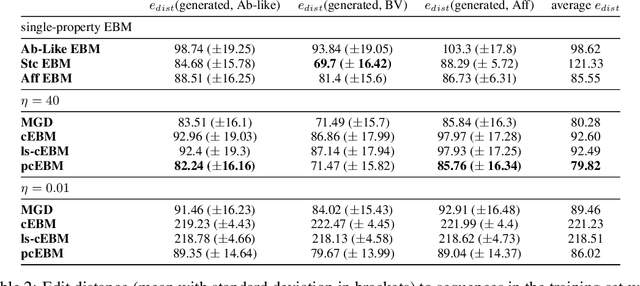
Deep generative models have emerged as a popular machine learning-based approach for inverse design problems in the life sciences. However, these problems often require sampling new designs that satisfy multiple properties of interest in addition to learning the data distribution. This multi-objective optimization becomes more challenging when properties are independent or orthogonal to each other. In this work, we propose a Pareto-compositional energy-based model (pcEBM), a framework that uses multiple gradient descent for sampling new designs that adhere to various constraints in optimizing distinct properties. We demonstrate its ability to learn non-convex Pareto fronts and generate sequences that simultaneously satisfy multiple desired properties across a series of real-world antibody design tasks.