 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTianle Ma
Incorporating Biological Knowledge with Factor Graph Neural Network for Interpretable Deep Learning
Jun 03, 2019
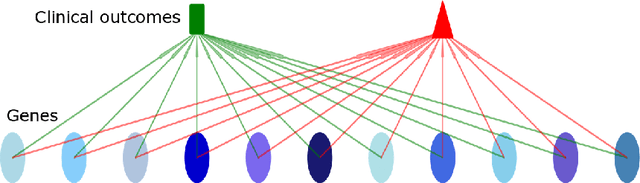
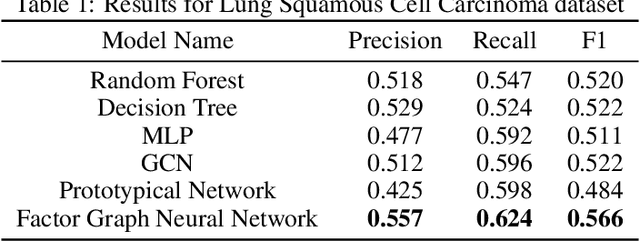
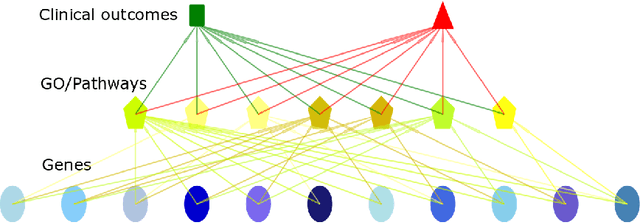
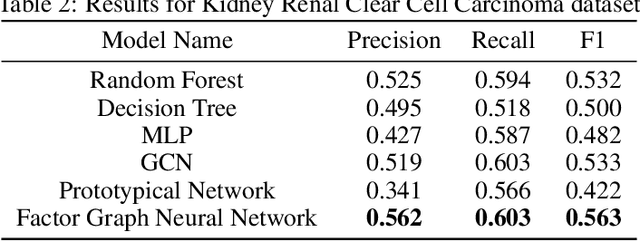
While deep learning has achieved great success in many fields, one common criticism about deep learning is its lack of interpretability. In most cases, the hidden units in a deep neural network do not have a clear semantic meaning or correspond to any physical entities. However, model interpretability and explainability are crucial in many biomedical applications. To address this challenge, we developed the Factor Graph Neural Network model that is interpretable and predictable by combining probabilistic graphical models with deep learning. We directly encode biological knowledge such as Gene Ontology as a factor graph into the model architecture, making the model transparent and interpretable. Furthermore, we devised an attention mechanism that can capture multi-scale hierarchical interactions among biological entities such as genes and Gene Ontology terms. With parameter sharing mechanism, the unrolled Factor Graph Neural Network model can be trained with stochastic depth and generalize well. We applied our model to two cancer genomic datasets to predict target clinical variables and achieved better results than other traditional machine learning and deep learning models. Our model can also be used for gene set enrichment analysis and selecting Gene Ontology terms that are important to target clinical variables.
Multi-view Factorization AutoEncoder with Network Constraints for Multi-omic Integrative Analysis
Sep 06, 2018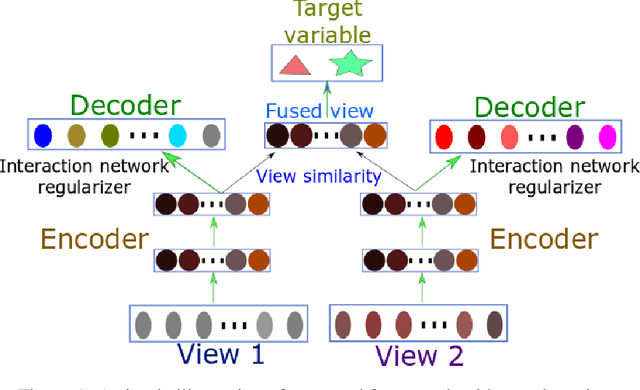

Multi-omic data provides multiple views of the same patients. Integrative analysis of multi-omic data is crucial to elucidate the molecular underpinning of disease etiology. However, multi-omic data has the "big p, small N" problem (the number of features is large, but the number of samples is small), it is challenging to train a complicated machine learning model from the multi-omic data alone and make it generalize well. Here we propose a framework termed Multi-view Factorization AutoEncoder with network constraints to integrate multi-omic data with domain knowledge (biological interactions networks). Our framework employs deep representation learning to learn feature embeddings and patient embeddings simultaneously, enabling us to integrate feature interaction network and patient view similarity network constraints into the training objective. The whole framework is end-to-end differentiable. We applied our approach to the TCGA Pan-cancer dataset and achieved satisfactory results to predict disease progression-free interval (PFI) and patient overall survival (OS) events. Code will be made publicly available.
AffinityNet: semi-supervised few-shot learning for disease type prediction
Sep 05, 2018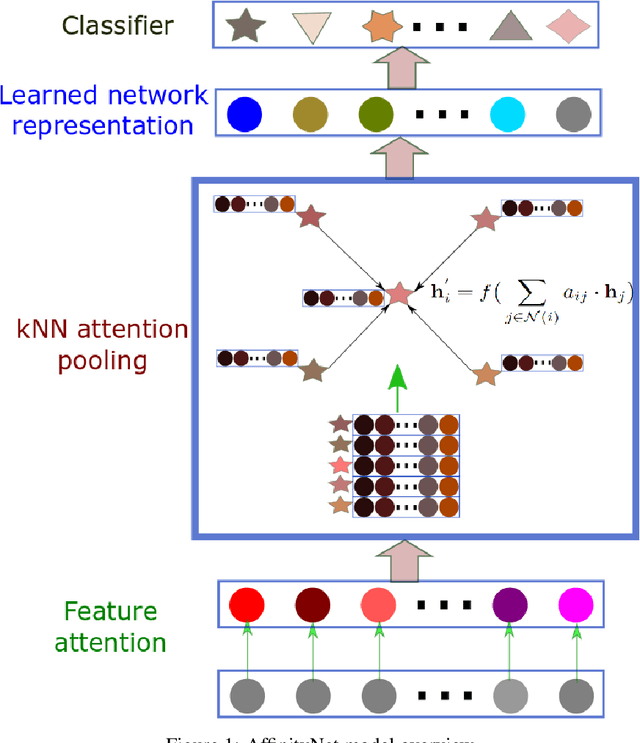

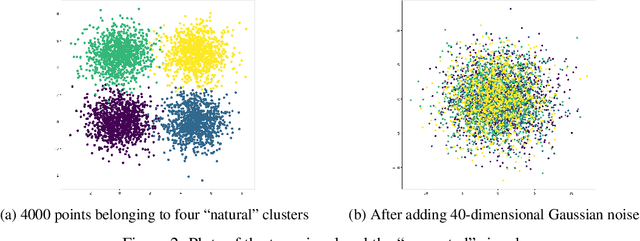
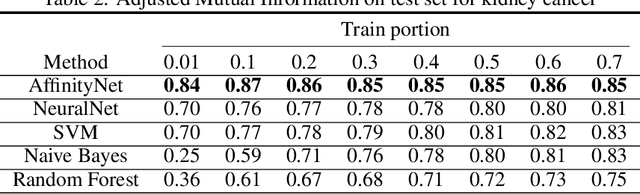
While deep learning has achieved great success in computer vision and many other fields, currently it does not work very well on patient genomic data with the "big p, small N" problem (i.e., a relatively small number of samples with high-dimensional features). In order to make deep learning work with a small amount of training data, we have to design new models that facilitate few-shot learning. Here we present the Affinity Network Model (AffinityNet), a data efficient deep learning model that can learn from a limited number of training examples and generalize well. The backbone of the AffinityNet model consists of stacked k-Nearest-Neighbor (kNN) attention pooling layers. The kNN attention pooling layer is a generalization of the Graph Attention Model (GAM), and can be applied to not only graphs but also any set of objects regardless of whether a graph is given or not. As a new deep learning module, kNN attention pooling layers can be plugged into any neural network model just like convolutional layers. As a simple special case of kNN attention pooling layer, feature attention layer can directly select important features that are useful for classification tasks. Experiments on both synthetic data and cancer genomic data from TCGA projects show that our AffinityNet model has better generalization power than conventional neural network models with little training data. The code is freely available at https://github.com/BeautyOfWeb/AffinityNet .