 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAnqi Liu
Density-Regression: Efficient and Distance-Aware Deep Regressor for Uncertainty Estimation under Distribution Shifts
Mar 07, 2024

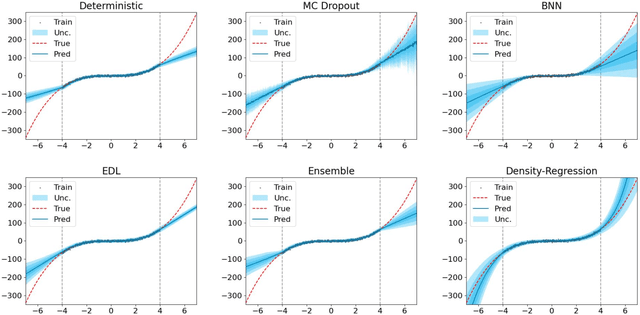

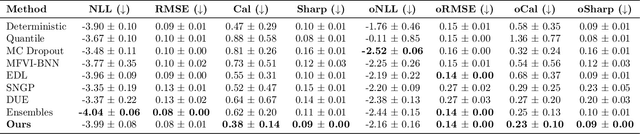
Morden deep ensembles technique achieves strong uncertainty estimation performance by going through multiple forward passes with different models. This is at the price of a high storage space and a slow speed in the inference (test) time. To address this issue, we propose Density-Regression, a method that leverages the density function in uncertainty estimation and achieves fast inference by a single forward pass. We prove it is distance aware on the feature space, which is a necessary condition for a neural network to produce high-quality uncertainty estimation under distribution shifts. Empirically, we conduct experiments on regression tasks with the cubic toy dataset, benchmark UCI, weather forecast with time series, and depth estimation under real-world shifted applications. We show that Density-Regression has competitive uncertainty estimation performance under distribution shifts with modern deep regressors while using a lower model size and a faster inference speed.
RORA: Robust Free-Text Rationale Evaluation
Mar 01, 2024Free-text rationales play a pivotal role in explainable NLP, bridging the knowledge and reasoning gaps behind a model's decision-making. However, due to the diversity of potential reasoning paths and a corresponding lack of definitive ground truth, their evaluation remains a challenge. Existing evaluation metrics rely on the degree to which a rationale supports a target label, but we find these fall short in evaluating rationales that inadvertently leak the labels. To address this problem, we propose RORA, a Robust free-text Rationale evaluation against label leakage. RORA quantifies the new information supplied by a rationale to justify the label. This is achieved by assessing the conditional V-information \citep{hewitt-etal-2021-conditional} with a predictive family robust against leaky features that can be exploited by a small model. RORA consistently outperforms existing approaches in evaluating human-written, synthetic, or model-generated rationales, particularly demonstrating robustness against label leakage. We also show that RORA aligns well with human judgment, providing a more reliable and accurate measurement across diverse free-text rationales.
Distributionally Robust Policy Evaluation under General Covariate Shift in Contextual Bandits
Jan 21, 2024We introduce a distributionally robust approach that enhances the reliability of offline policy evaluation in contextual bandits under general covariate shifts. Our method aims to deliver robust policy evaluation results in the presence of discrepancies in both context and policy distribution between logging and target data. Central to our methodology is the application of robust regression, a distributionally robust technique tailored here to improve the estimation of conditional reward distribution from logging data. Utilizing the reward model obtained from robust regression, we develop a comprehensive suite of policy value estimators, by integrating our reward model into established evaluation frameworks, namely direct methods and doubly robust methods. Through theoretical analysis, we further establish that the proposed policy value estimators offer a finite sample upper bound for the bias, providing a clear advantage over traditional methods, especially when the shift is large. Finally, we designed an extensive range of policy evaluation scenarios, covering diverse magnitudes of shifts and a spectrum of logging and target policies. Our empirical results indicate that our approach significantly outperforms baseline methods, most notably in 90% of the cases under the policy shift-only settings and 72% of the scenarios under the general covariate shift settings.
Multi-View Variational Autoencoder for Missing Value Imputation in Untargeted Metabolomics
Oct 12, 2023
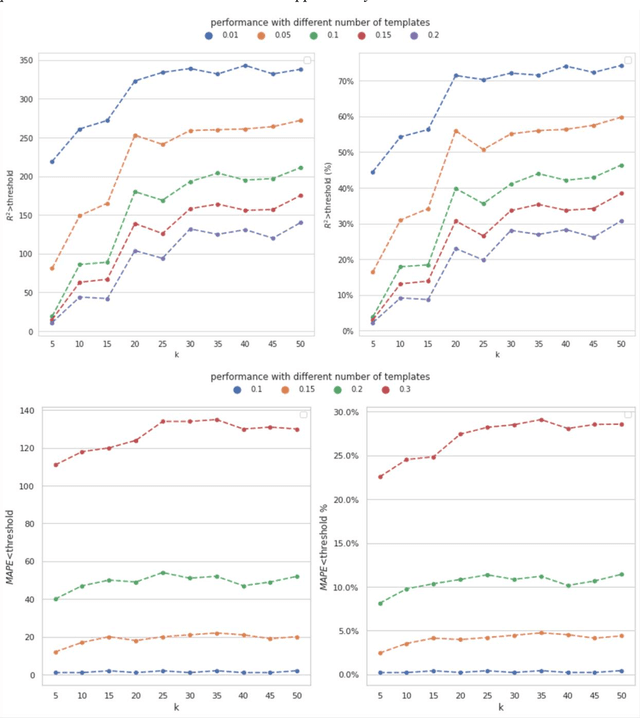
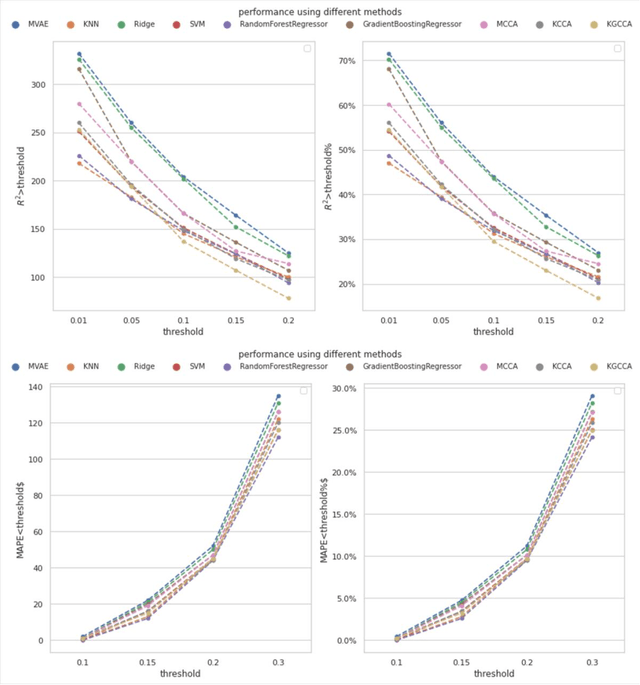
Background: Missing data is a common challenge in mass spectrometry-based metabolomics, which can lead to biased and incomplete analyses. The integration of whole-genome sequencing (WGS) data with metabolomics data has emerged as a promising approach to enhance the accuracy of data imputation in metabolomics studies. Method: In this study, we propose a novel method that leverages the information from WGS data and reference metabolites to impute unknown metabolites. Our approach utilizes a multi-view variational autoencoder to jointly model the burden score, polygenetic risk score (PGS), and linkage disequilibrium (LD) pruned single nucleotide polymorphisms (SNPs) for feature extraction and missing metabolomics data imputation. By learning the latent representations of both omics data, our method can effectively impute missing metabolomics values based on genomic information. Results: We evaluate the performance of our method on empirical metabolomics datasets with missing values and demonstrate its superiority compared to conventional imputation techniques. Using 35 template metabolites derived burden scores, PGS and LD-pruned SNPs, the proposed methods achieved r2-scores > 0.01 for 71.55% of metabolites. Conclusion: The integration of WGS data in metabolomics imputation not only improves data completeness but also enhances downstream analyses, paving the way for more comprehensive and accurate investigations of metabolic pathways and disease associations. Our findings offer valuable insights into the potential benefits of utilizing WGS data for metabolomics data imputation and underscore the importance of leveraging multi-modal data integration in precision medicine research.
MegaWika: Millions of reports and their sources across 50 diverse languages
Jul 13, 2023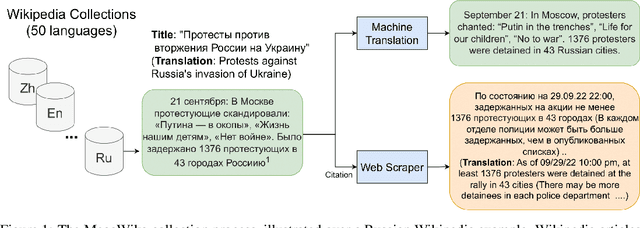
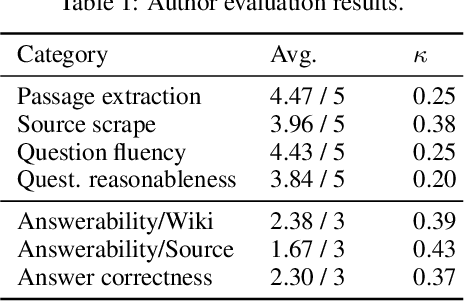
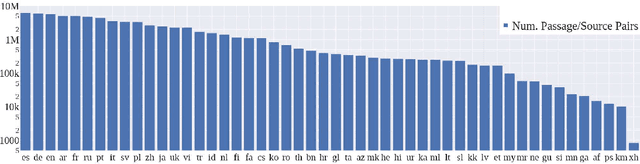
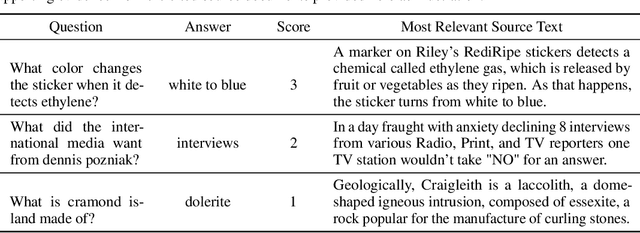
To foster the development of new models for collaborative AI-assisted report generation, we introduce MegaWika, consisting of 13 million Wikipedia articles in 50 diverse languages, along with their 71 million referenced source materials. We process this dataset for a myriad of applications, going beyond the initial Wikipedia citation extraction and web scraping of content, including translating non-English articles for cross-lingual applications and providing FrameNet parses for automated semantic analysis. MegaWika is the largest resource for sentence-level report generation and the only report generation dataset that is multilingual. We manually analyze the quality of this resource through a semantically stratified sample. Finally, we provide baseline results and trained models for crucial steps in automated report generation: cross-lingual question answering and citation retrieval.
Double-Weighting for Covariate Shift Adaptation
May 15, 2023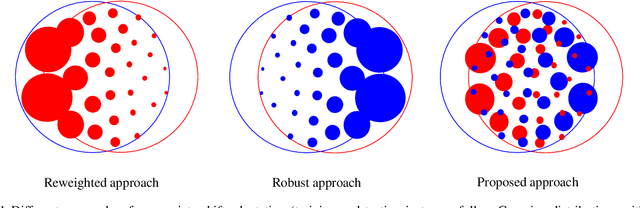

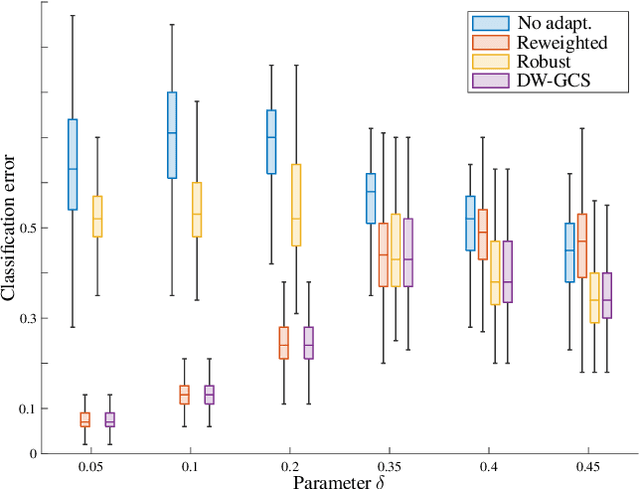
Supervised learning is often affected by a covariate shift in which the marginal distributions of instances (covariates $x$) of training and testing samples $\mathrm{p}_\text{tr}(x)$ and $\mathrm{p}_\text{te}(x)$ are different but the label conditionals coincide. Existing approaches address such covariate shift by either using the ratio $\mathrm{p}_\text{te}(x)/\mathrm{p}_\text{tr}(x)$ to weight training samples (reweighting methods) or using the ratio $\mathrm{p}_\text{tr}(x)/\mathrm{p}_\text{te}(x)$ to weight testing samples (robust methods). However, the performance of such approaches can be poor under support mismatch or when the above ratios take large values. We propose a minimax risk classification (MRC) approach for covariate shift adaptation that avoids such limitations by weighting both training and testing samples. In addition, we develop effective techniques that obtain both sets of weights and generalize the conventional kernel mean matching method. We provide novel generalization bounds for our method that show a significant increase in the effective sample size compared with reweighted methods. The proposed method also achieves enhanced classification performance in both synthetic and empirical experiments.
CLCLSA: Cross-omics Linked embedding with Contrastive Learning and Self Attention for multi-omics integration with incomplete multi-omics data
Apr 12, 2023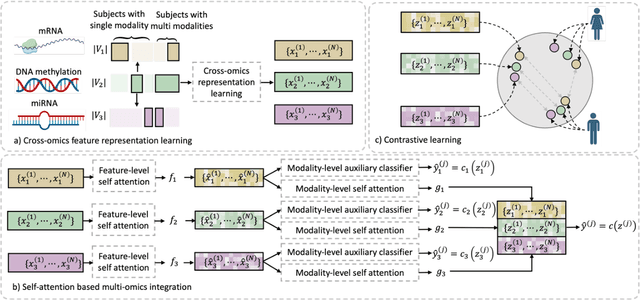
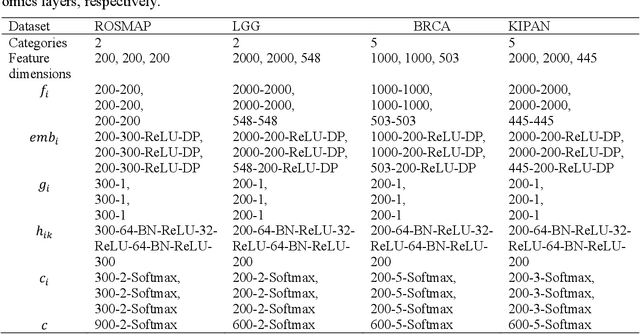
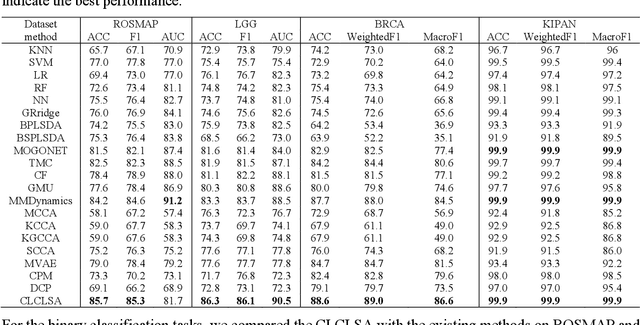
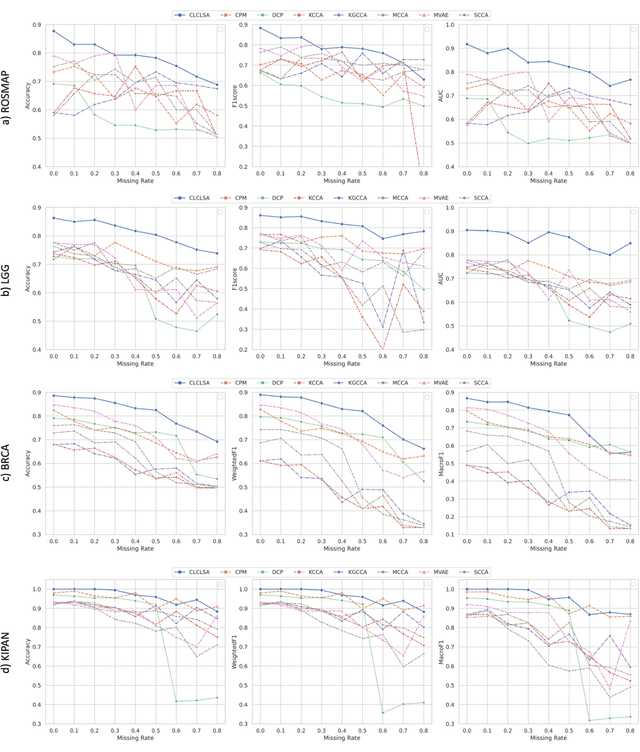
Integration of heterogeneous and high-dimensional multi-omics data is becoming increasingly important in understanding genetic data. Each omics technique only provides a limited view of the underlying biological process and integrating heterogeneous omics layers simultaneously would lead to a more comprehensive and detailed understanding of diseases and phenotypes. However, one obstacle faced when performing multi-omics data integration is the existence of unpaired multi-omics data due to instrument sensitivity and cost. Studies may fail if certain aspects of the subjects are missing or incomplete. In this paper, we propose a deep learning method for multi-omics integration with incomplete data by Cross-omics Linked unified embedding with Contrastive Learning and Self Attention (CLCLSA). Utilizing complete multi-omics data as supervision, the model employs cross-omics autoencoders to learn the feature representation across different types of biological data. The multi-omics contrastive learning, which is used to maximize the mutual information between different types of omics, is employed before latent feature concatenation. In addition, the feature-level self-attention and omics-level self-attention are employed to dynamically identify the most informative features for multi-omics data integration. Extensive experiments were conducted on four public multi-omics datasets. The experimental results indicated that the proposed CLCLSA outperformed the state-of-the-art approaches for multi-omics data classification using incomplete multi-omics data.
Density-Softmax: Scalable and Distance-Aware Uncertainty Estimation under Distribution Shifts
Feb 13, 2023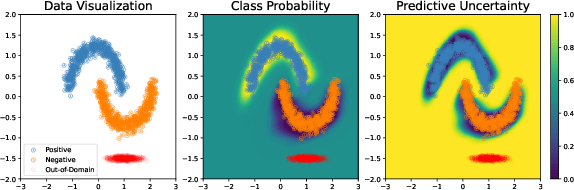
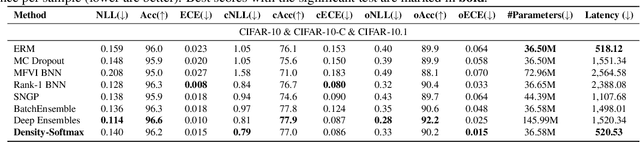


Prevalent deep learning models suffer from significant over-confidence under distribution shifts. In this paper, we propose Density-Softmax, a single deterministic approach for uncertainty estimation via a combination of density function with the softmax layer. By using the latent representation's likelihood value, our approach produces more uncertain predictions when test samples are distant from the training samples. Theoretically, we prove that Density-Softmax is distance aware, which means its associated uncertainty metrics are monotonic functions of distance metrics. This has been shown to be a necessary condition for a neural network to produce high-quality uncertainty estimation. Empirically, our method enjoys similar computational efficiency as standard softmax on shifted CIFAR-10, CIFAR-100, and ImageNet dataset across modern deep learning architectures. Notably, Density-Softmax uses 4 times fewer parameters than Deep Ensembles and 6 times lower latency than Rank-1 Bayesian Neural Network, while obtaining competitive predictive performance and lower calibration errors under distribution shifts.
ImageNomer: developing an fMRI and omics visualization tool to detect racial bias in functional connectivity
Feb 01, 2023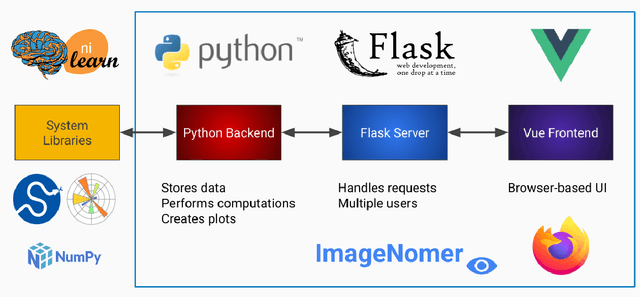
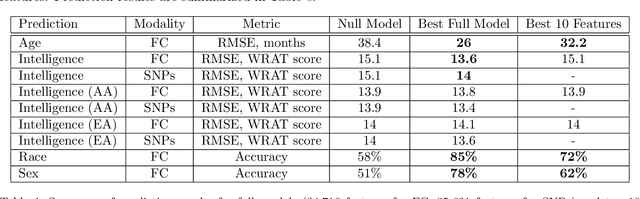
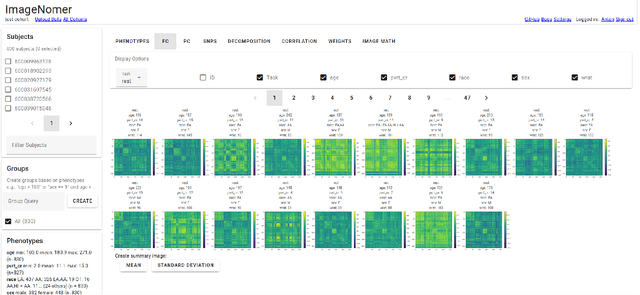
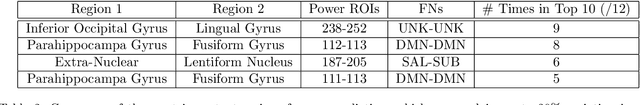
It can be difficult to identify trends and perform quality control in large, high-dimensional fMRI or omics datasets. To remedy this, we develop ImageNomer, a data visualization and analysis tool that allows inspection of both subject-level and cohort-level features. The tool allows visualization of phenotype correlation with functional connectivity (FC), partial connectivity (PC), dictionary components (PCA and our own method), and genomic data (single-nucleotide polymorphisms, SNPs). In addition, it allows visualization of weights from arbitrary ML models. ImageNomer is built with a Python backend and a Vue frontend. We validate ImageNomer using the Philadelphia Neurodevelopmental Cohort (PNC) dataset, which contains multitask fMRI and SNP data of healthy adolescents. Using correlation, greedy selection, or model weights, we find that a set of 10 FC features can explain 15% of variation in age, compared to 35% for the full 34,716 feature model. The four most significant FCs are either between bilateral default mode network (DMN) regions or spatially proximal subcortical areas. Additionally, we show that whereas both FC (fMRI) and SNPs (genomic) features can account for 10-15% of intelligence variation, this predictive ability disappears when controlling for race. We find that FC features can be used to predict race with 85% accuracy, compared to 78% accuracy for sex prediction. Using ImageNomer, this work casts doubt on the possibility of finding unbiased intelligence-related features in fMRI and SNPs of healthy adolescents.
Discovering Customer-Service Dialog System with Semi-Supervised Learning and Coarse-to-Fine Intent Detection
Dec 23, 2022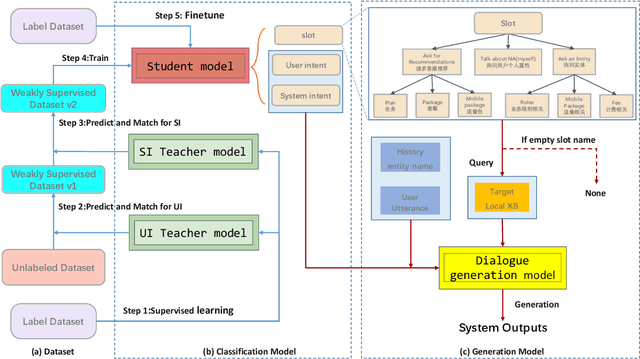


Task-oriented dialog(TOD) aims to assist users in achieving specific goals through multi-turn conversation. Recently, good results have been obtained based on large pre-trained models. However, the labeled-data scarcity hinders the efficient development of TOD systems at scale. In this work, we constructed a weakly supervised dataset based on a teacher/student paradigm that leverages a large collection of unlabelled dialogues. Furthermore, we built a modular dialogue system and integrated coarse-to-fine grained classification for user intent detection. Experiments show that our method can reach the dialog goal with a higher success rate and generate more coherent responses.