 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFrauke Wilm
Style-Extracting Diffusion Models for Semi-Supervised Histopathology Segmentation
Mar 21, 2024
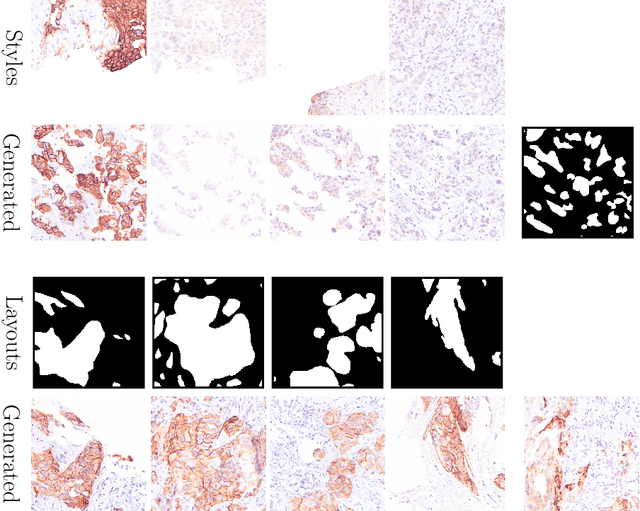
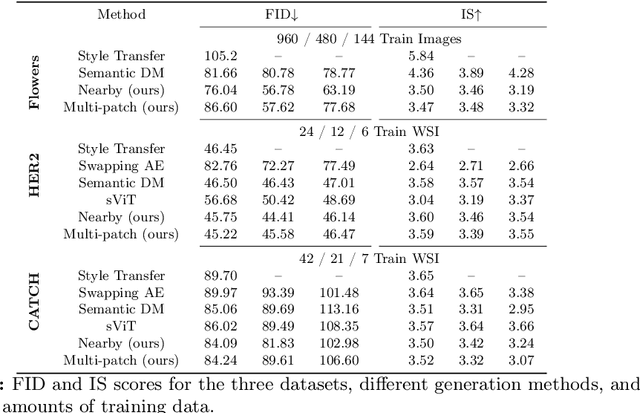
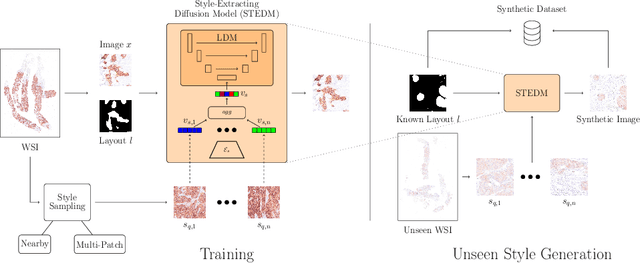
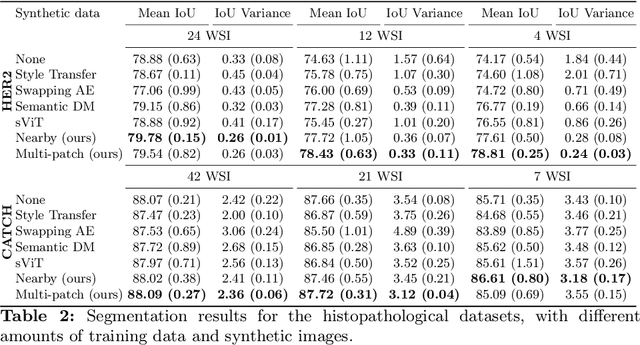
Deep learning-based image generation has seen significant advancements with diffusion models, notably improving the quality of generated images. Despite these developments, generating images with unseen characteristics beneficial for downstream tasks has received limited attention. To bridge this gap, we propose Style-Extracting Diffusion Models, featuring two conditioning mechanisms. Specifically, we utilize 1) a style conditioning mechanism which allows to inject style information of previously unseen images during image generation and 2) a content conditioning which can be targeted to a downstream task, e.g., layout for segmentation. We introduce a trainable style encoder to extract style information from images, and an aggregation block that merges style information from multiple style inputs. This architecture enables the generation of images with unseen styles in a zero-shot manner, by leveraging styles from unseen images, resulting in more diverse generations. In this work, we use the image layout as target condition and first show the capability of our method on a natural image dataset as a proof-of-concept. We further demonstrate its versatility in histopathology, where we combine prior knowledge about tissue composition and unannotated data to create diverse synthetic images with known layouts. This allows us to generate additional synthetic data to train a segmentation network in a semi-supervised fashion. We verify the added value of the generated images by showing improved segmentation results and lower performance variability between patients when synthetic images are included during segmentation training. Our code will be made publicly available at [LINK].
Analysing Diffusion Segmentation for Medical Images
Mar 21, 2024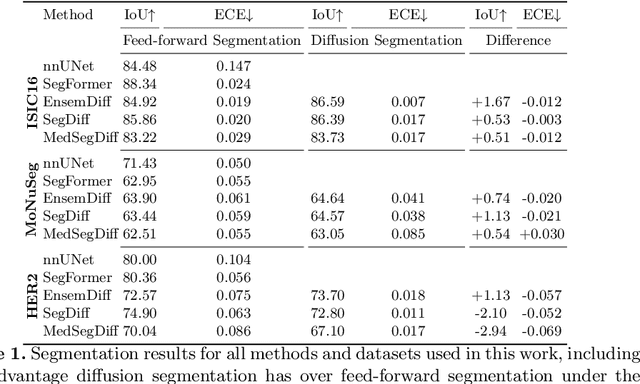
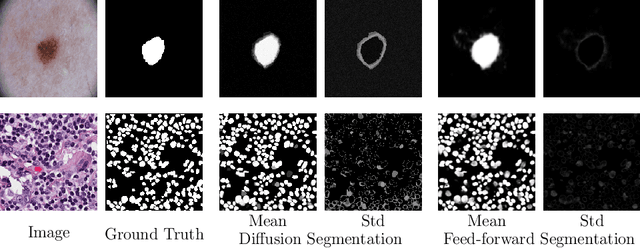
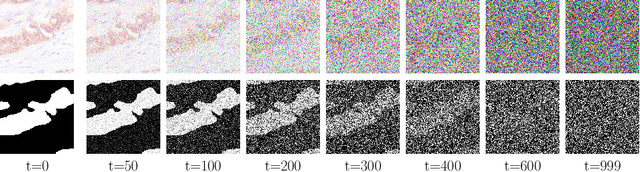
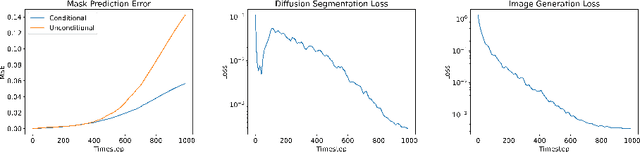
Denoising Diffusion Probabilistic models have become increasingly popular due to their ability to offer probabilistic modeling and generate diverse outputs. This versatility inspired their adaptation for image segmentation, where multiple predictions of the model can produce segmentation results that not only achieve high quality but also capture the uncertainty inherent in the model. Here, powerful architectures were proposed for improving diffusion segmentation performance. However, there is a notable lack of analysis and discussions on the differences between diffusion segmentation and image generation, and thorough evaluations are missing that distinguish the improvements these architectures provide for segmentation in general from their benefit for diffusion segmentation specifically. In this work, we critically analyse and discuss how diffusion segmentation for medical images differs from diffusion image generation, with a particular focus on the training behavior. Furthermore, we conduct an assessment how proposed diffusion segmentation architectures perform when trained directly for segmentation. Lastly, we explore how different medical segmentation tasks influence the diffusion segmentation behavior and the diffusion process could be adapted accordingly. With these analyses, we aim to provide in-depth insights into the behavior of diffusion segmentation that allow for a better design and evaluation of diffusion segmentation methods in the future.
Rethinking U-net Skip Connections for Biomedical Image Segmentation
Feb 13, 2024The U-net architecture has significantly impacted deep learning-based segmentation of medical images. Through the integration of long-range skip connections, it facilitated the preservation of high-resolution features. Out-of-distribution data can, however, substantially impede the performance of neural networks. Previous works showed that the trained network layers differ in their susceptibility to this domain shift, e.g., shallow layers are more affected than deeper layers. In this work, we investigate the implications of this observation of layer sensitivity to domain shifts of U-net-style segmentation networks. By copying features of shallow layers to corresponding decoder blocks, these bear the risk of re-introducing domain-specific information. We used a synthetic dataset to model different levels of data distribution shifts and evaluated the impact on downstream segmentation performance. We quantified the inherent domain susceptibility of each network layer, using the Hellinger distance. These experiments confirmed the higher domain susceptibility of earlier network layers. When gradually removing skip connections, a decrease in domain susceptibility of deeper layers could be observed. For downstream segmentation performance, the original U-net outperformed the variant without any skip connections. The best performance, however, was achieved when removing the uppermost skip connection - not only in the presence of domain shifts but also for in-domain test data. We validated our results on three clinical datasets - two histopathology datasets and one magnetic resonance dataset - with performance increases of up to 10% in-domain and 13% cross-domain when removing the uppermost skip connection.
Domain generalization across tumor types, laboratories, and species -- insights from the 2022 edition of the Mitosis Domain Generalization Challenge
Sep 27, 2023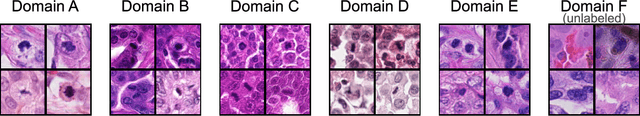
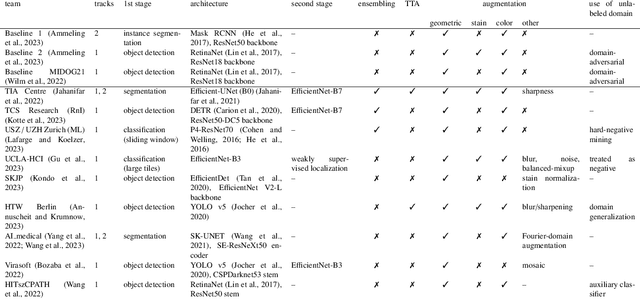
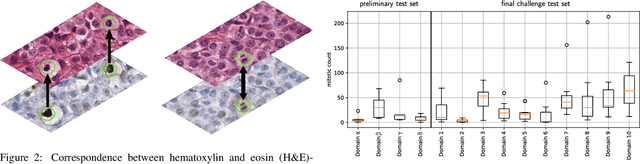
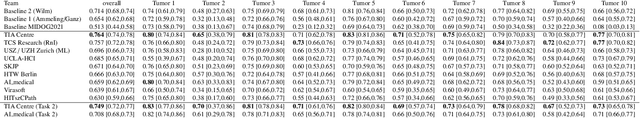
Recognition of mitotic figures in histologic tumor specimens is highly relevant to patient outcome assessment. This task is challenging for algorithms and human experts alike, with deterioration of algorithmic performance under shifts in image representations. Considerable covariate shifts occur when assessment is performed on different tumor types, images are acquired using different digitization devices, or specimens are produced in different laboratories. This observation motivated the inception of the 2022 challenge on MItosis Domain Generalization (MIDOG 2022). The challenge provided annotated histologic tumor images from six different domains and evaluated the algorithmic approaches for mitotic figure detection provided by nine challenge participants on ten independent domains. Ground truth for mitotic figure detection was established in two ways: a three-expert consensus and an independent, immunohistochemistry-assisted set of labels. This work represents an overview of the challenge tasks, the algorithmic strategies employed by the participants, and potential factors contributing to their success. With an $F_1$ score of 0.764 for the top-performing team, we summarize that domain generalization across various tumor domains is possible with today's deep learning-based recognition pipelines. When assessed against the immunohistochemistry-assisted reference standard, all methods resulted in reduced recall scores, but with only minor changes in the order of participants in the ranking.
Adaptive Region Selection for Active Learning in Whole Slide Image Semantic Segmentation
Jul 14, 2023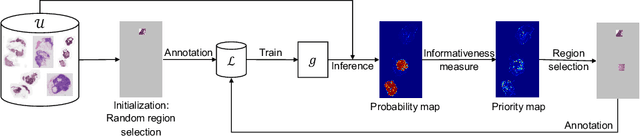
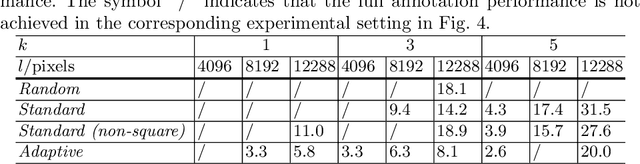
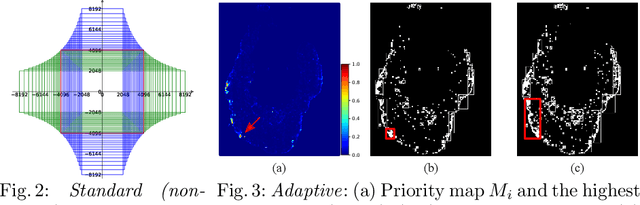
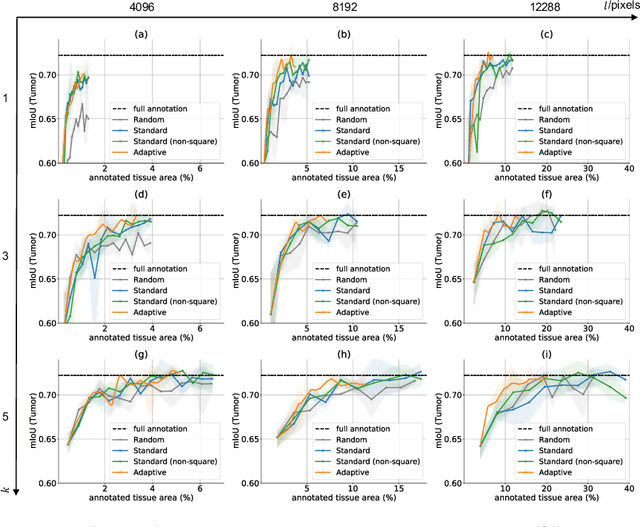
The process of annotating histological gigapixel-sized whole slide images (WSIs) at the pixel level for the purpose of training a supervised segmentation model is time-consuming. Region-based active learning (AL) involves training the model on a limited number of annotated image regions instead of requesting annotations of the entire images. These annotation regions are iteratively selected, with the goal of optimizing model performance while minimizing the annotated area. The standard method for region selection evaluates the informativeness of all square regions of a specified size and then selects a specific quantity of the most informative regions. We find that the efficiency of this method highly depends on the choice of AL step size (i.e., the combination of region size and the number of selected regions per WSI), and a suboptimal AL step size can result in redundant annotation requests or inflated computation costs. This paper introduces a novel technique for selecting annotation regions adaptively, mitigating the reliance on this AL hyperparameter. Specifically, we dynamically determine each region by first identifying an informative area and then detecting its optimal bounding box, as opposed to selecting regions of a uniform predefined shape and size as in the standard method. We evaluate our method using the task of breast cancer metastases segmentation on the public CAMELYON16 dataset and show that it consistently achieves higher sampling efficiency than the standard method across various AL step sizes. With only 2.6\% of tissue area annotated, we achieve full annotation performance and thereby substantially reduce the costs of annotating a WSI dataset. The source code is available at https://github.com/DeepMicroscopy/AdaptiveRegionSelection.
Multi-Scanner Canine Cutaneous Squamous Cell Carcinoma Histopathology Dataset
Jan 11, 2023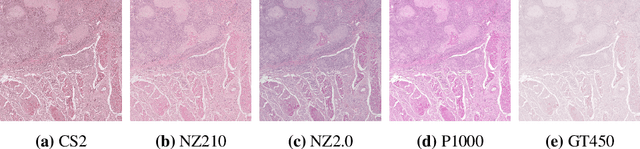
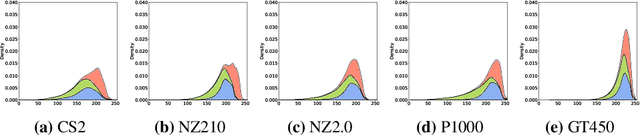
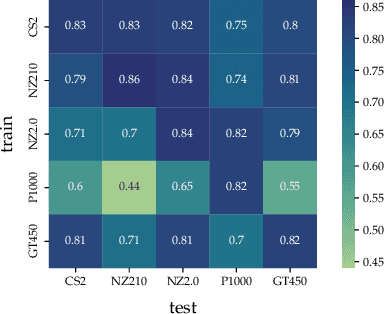
In histopathology, scanner-induced domain shifts are known to impede the performance of trained neural networks when tested on unseen data. Multi-domain pre-training or dedicated domain-generalization techniques can help to develop domain-agnostic algorithms. For this, multi-scanner datasets with a high variety of slide scanning systems are highly desirable. We present a publicly available multi-scanner dataset of canine cutaneous squamous cell carcinoma histopathology images, composed of 44 samples digitized with five slide scanners. This dataset provides local correspondences between images and thereby isolates the scanner-induced domain shift from other inherent, e.g. morphology-induced domain shifts. To highlight scanner differences, we present a detailed evaluation of color distributions, sharpness, and contrast of the individual scanner subsets. Additionally, to quantify the inherent scanner-induced domain shift, we train a tumor segmentation network on each scanner subset and evaluate the performance both in- and cross-domain. We achieve a class-averaged in-domain intersection over union coefficient of up to 0.86 and observe a cross-domain performance decrease of up to 0.38, which confirms the inherent domain shift of the presented dataset and its negative impact on the performance of deep neural networks.
Mind the Gap: Scanner-induced domain shifts pose challenges for representation learning in histopathology
Nov 29, 2022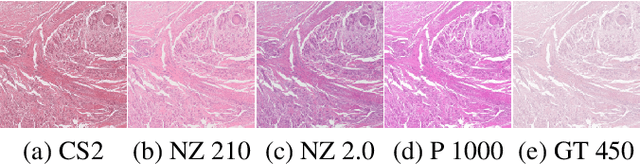
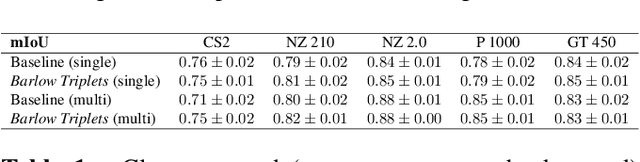
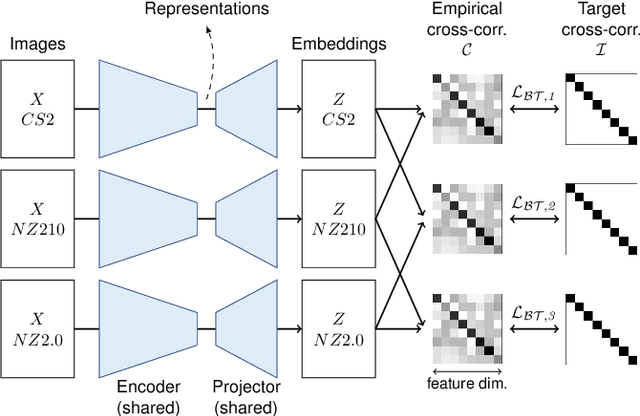
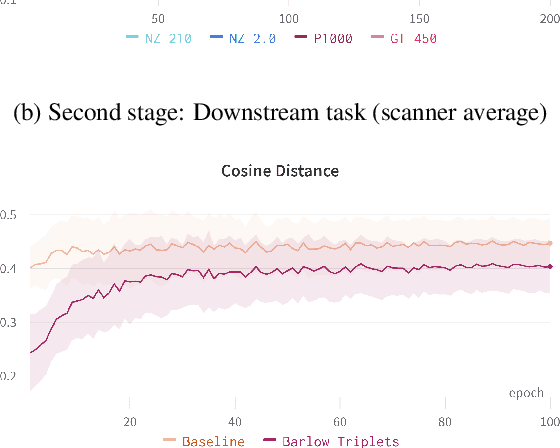
Computer-aided systems in histopathology are often challenged by various sources of domain shift that impact the performance of these algorithms considerably. We investigated the potential of using self-supervised pre-training to overcome scanner-induced domain shifts for the downstream task of tumor segmentation. For this, we present the Barlow Triplets to learn scanner-invariant representations from a multi-scanner dataset with local image correspondences. We show that self-supervised pre-training successfully aligned different scanner representations, which, interestingly only results in a limited benefit for our downstream task. We thereby provide insights into the influence of scanner characteristics for downstream applications and contribute to a better understanding of why established self-supervised methods have not yet shown the same success on histopathology data as they have for natural images.
Improved HER2 Tumor Segmentation with Subtype Balancing using Deep Generative Networks
Nov 11, 2022



Tumor segmentation in histopathology images is often complicated by its composition of different histological subtypes and class imbalance. Oversampling subtypes with low prevalence features is not a satisfactory solution since it eventually leads to overfitting. We propose to create synthetic images with semantically-conditioned deep generative networks and to combine subtype-balanced synthetic images with the original dataset to achieve better segmentation performance. We show the suitability of Generative Adversarial Networks (GANs) and especially diffusion models to create realistic images based on subtype-conditioning for the use case of HER2-stained histopathology. Additionally, we show the capability of diffusion models to conditionally inpaint HER2 tumor areas with modified subtypes. Combining the original dataset with the same amount of diffusion-generated images increased the tumor Dice score from 0.833 to 0.854 and almost halved the variance between the HER2 subtype recalls. These results create the basis for more reliable automatic HER2 analysis with lower performance variance between individual HER2 subtypes.
Mitosis domain generalization in histopathology images -- The MIDOG challenge
Apr 06, 2022
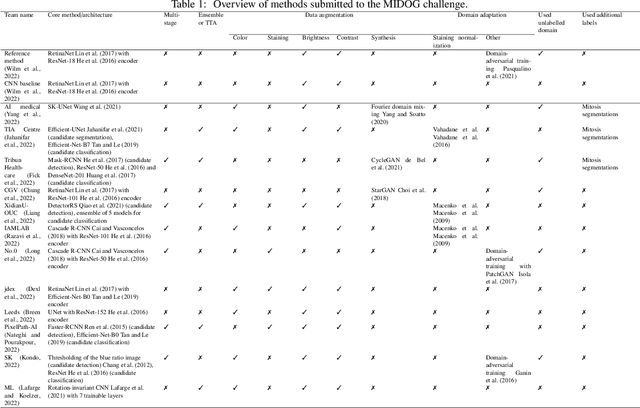
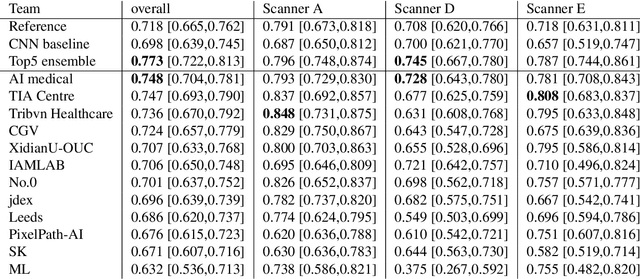
The density of mitotic figures within tumor tissue is known to be highly correlated with tumor proliferation and thus is an important marker in tumor grading. Recognition of mitotic figures by pathologists is known to be subject to a strong inter-rater bias, which limits the prognostic value. State-of-the-art deep learning methods can support the expert in this assessment but are known to strongly deteriorate when applied in a different clinical environment than was used for training. One decisive component in the underlying domain shift has been identified as the variability caused by using different whole slide scanners. The goal of the MICCAI MIDOG 2021 challenge has been to propose and evaluate methods that counter this domain shift and derive scanner-agnostic mitosis detection algorithms. The challenge used a training set of 200 cases, split across four scanning systems. As a test set, an additional 100 cases split across four scanning systems, including two previously unseen scanners, were given. The best approaches performed on an expert level, with the winning algorithm yielding an F_1 score of 0.748 (CI95: 0.704-0.781). In this paper, we evaluate and compare the approaches that were submitted to the challenge and identify methodological factors contributing to better performance.
Pan-Tumor CAnine cuTaneous Cancer Histology (CATCH) Dataset
Jan 27, 2022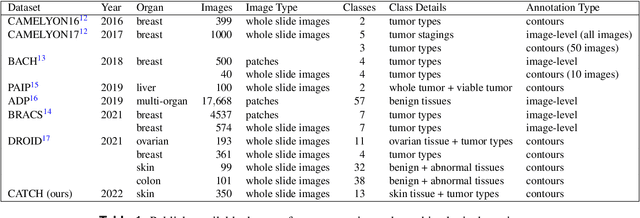
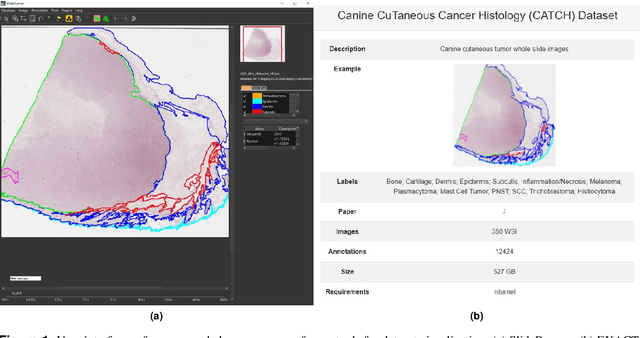
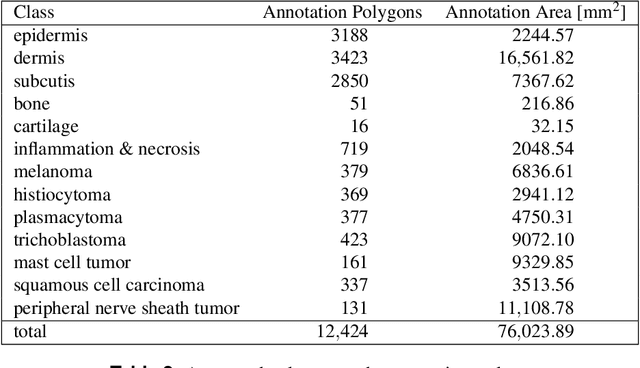

Due to morphological similarities, the differentiation of histologic sections of cutaneous tumors into individual subtypes can be challenging. Recently, deep learning-based approaches have proven their potential for supporting pathologists in this regard. However, many of these supervised algorithms require a large amount of annotated data for robust development. We present a publicly available dataset consisting of 350 whole slide images of seven different canine cutaneous tumors complemented by 12,424 polygon annotations for 13 histologic classes including seven cutaneous tumor subtypes. Regarding sample size and annotation extent, this exceeds most publicly available datasets which are oftentimes limited to the tumor area or merely provide patch-level annotations. We validated our model for tissue segmentation, achieving a class-averaged Jaccard coefficient of 0.7047, and 0.9044 for tumor in particular. For tumor subtype classification, we achieve a slide-level accuracy of 0.9857. Since canine cutaneous tumors possess various histologic homologies to human tumors, we believe that the added value of this dataset is not limited to veterinary pathology but extends to more general fields of application.