 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeJialu Wu
Generative AI for Controllable Protein Sequence Design: A Survey
Feb 16, 2024
The design of novel protein sequences with targeted functionalities underpins a central theme in protein engineering, impacting diverse fields such as drug discovery and enzymatic engineering. However, navigating this vast combinatorial search space remains a severe challenge due to time and financial constraints. This scenario is rapidly evolving as the transformative advancements in AI, particularly in the realm of generative models and optimization algorithms, have been propelling the protein design field towards an unprecedented revolution. In this survey, we systematically review recent advances in generative AI for controllable protein sequence design. To set the stage, we first outline the foundational tasks in protein sequence design in terms of the constraints involved and present key generative models and optimization algorithms. We then offer in-depth reviews of each design task and discuss the pertinent applications. Finally, we identify the unresolved challenges and highlight research opportunities that merit deeper exploration.
From molecules to scaffolds to functional groups: building context-dependent molecular representation via multi-channel learning
Nov 05, 2023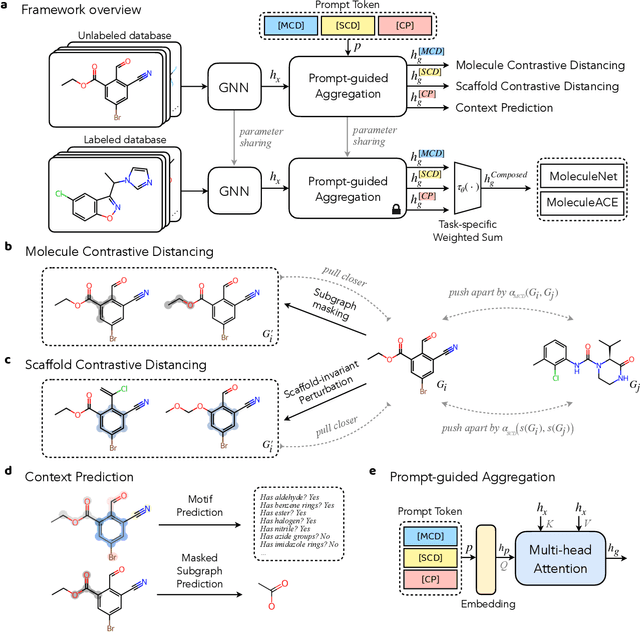
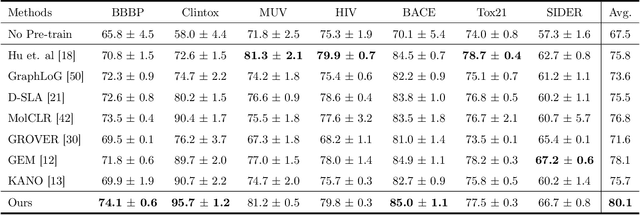
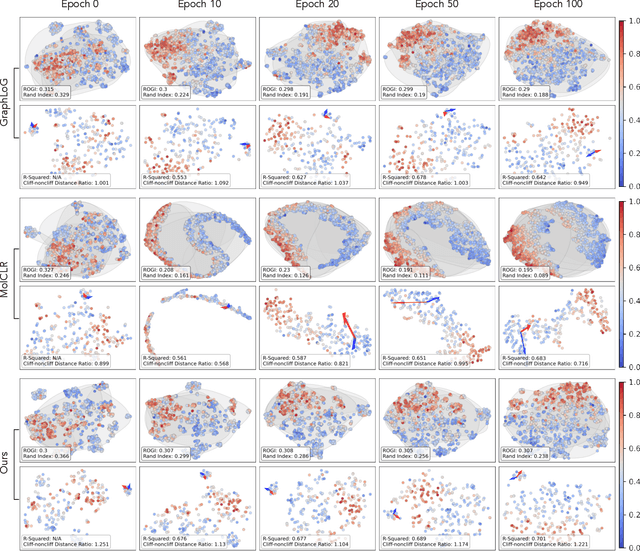
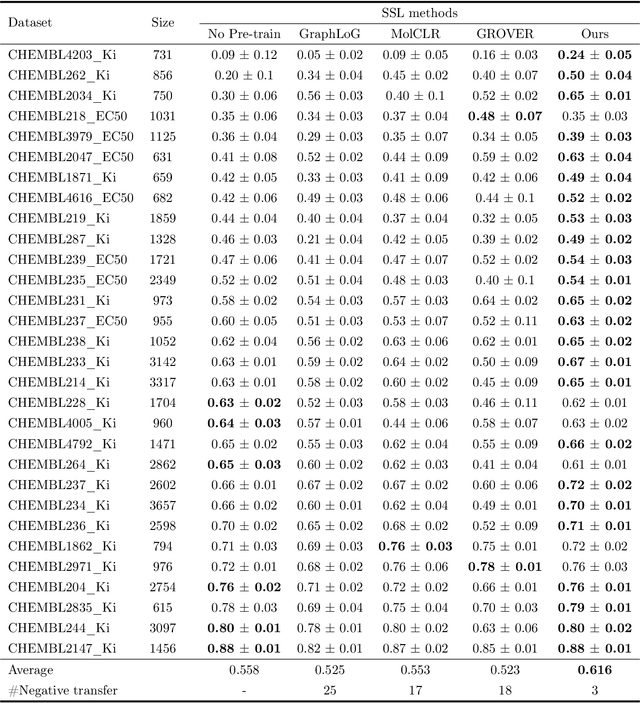
Reliable molecular property prediction is essential for various scientific endeavors and industrial applications, such as drug discovery. However, the scarcity of data, combined with the highly non-linear causal relationships between physicochemical and biological properties and conventional molecular featurization schemes, complicates the development of robust molecular machine learning models. Self-supervised learning (SSL) has emerged as a popular solution, utilizing large-scale, unannotated molecular data to learn a foundational representation of chemical space that might be advantageous for downstream tasks. Yet, existing molecular SSL methods largely overlook domain-specific knowledge, such as molecular similarity and scaffold importance, as well as the context of the target application when operating over the large chemical space. This paper introduces a novel learning framework that leverages the knowledge of structural hierarchies within molecular structures, embeds them through separate pre-training tasks over distinct channels, and employs a task-specific channel selection to compose a context-dependent representation. Our approach demonstrates competitive performance across various molecular property benchmarks and establishes some state-of-the-art results. It further offers unprecedented advantages in particularly challenging yet ubiquitous scenarios like activity cliffs with enhanced robustness and generalizability compared to other baselines.
MolHF: A Hierarchical Normalizing Flow for Molecular Graph Generation
May 15, 2023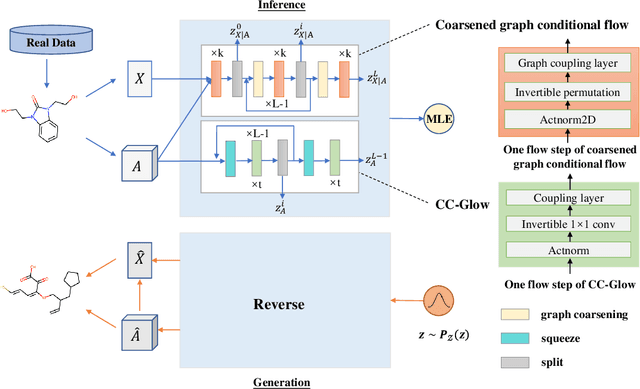
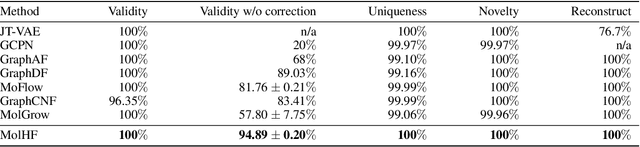
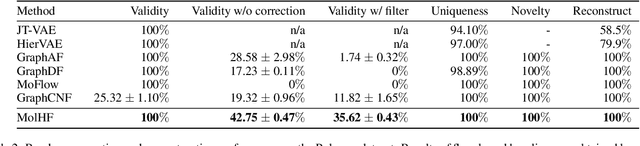
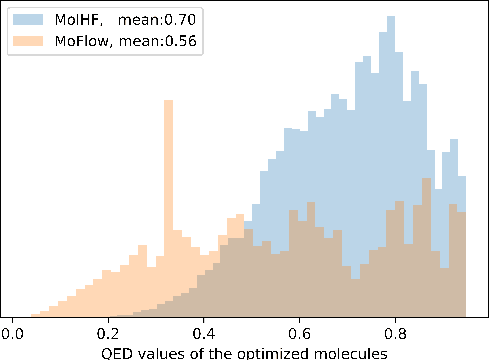
Molecular de novo design is a critical yet challenging task in scientific fields, aiming to design novel molecular structures with desired property profiles. Significant progress has been made by resorting to generative models for graphs. However, limited attention is paid to hierarchical generative models, which can exploit the inherent hierarchical structure (with rich semantic information) of the molecular graphs and generate complex molecules of larger size that we shall demonstrate to be difficult for most existing models. The primary challenge to hierarchical generation is the non-differentiable issue caused by the generation of intermediate discrete coarsened graph structures. To sidestep this issue, we cast the tricky hierarchical generation problem over discrete spaces as the reverse process of hierarchical representation learning and propose MolHF, a new hierarchical flow-based model that generates molecular graphs in a coarse-to-fine manner. Specifically, MolHF first generates bonds through a multi-scale architecture, then generates atoms based on the coarsened graph structure at each scale. We demonstrate that MolHF achieves state-of-the-art performance in random generation and property optimization, implying its high capacity to model data distribution. Furthermore, MolHF is the first flow-based model that can be applied to model larger molecules (polymer) with more than 100 heavy atoms. The code and models are available at https://github.com/violet-sto/MolHF.
Sample-efficient Multi-objective Molecular Optimization with GFlowNets
Feb 08, 2023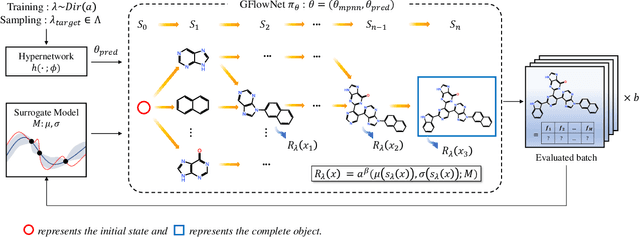
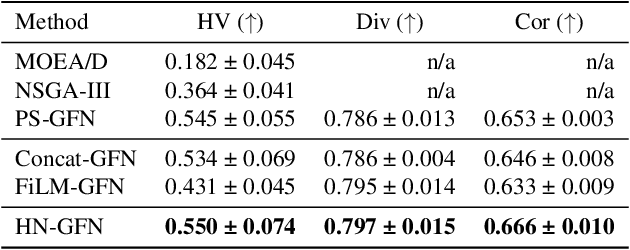
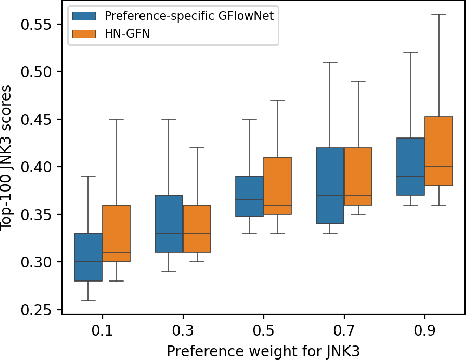
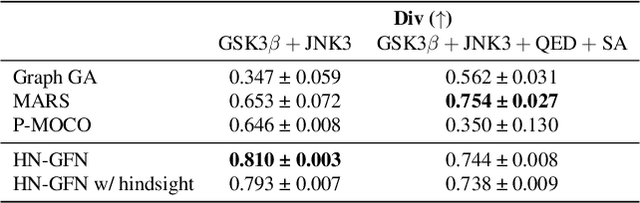
Many crucial scientific problems involve designing novel molecules with desired properties, which can be formulated as an expensive black-box optimization problem over the discrete chemical space. Computational methods have achieved initial success but still struggle with simultaneously optimizing multiple competing properties in a sample-efficient manner. In this work, we propose a multi-objective Bayesian optimization (MOBO) algorithm leveraging the hypernetwork-based GFlowNets (HN-GFN) as an acquisition function optimizer, with the purpose of sampling a diverse batch of candidate molecular graphs from an approximate Pareto front. Using a single preference-conditioned hypernetwork, HN-GFN learns to explore various trade-offs between objectives. Inspired by reinforcement learning, we further propose a hindsight-like off-policy strategy to share high-performing molecules among different preferences in order to speed up learning for HN-GFN. Through synthetic experiments, we illustrate that HN-GFN has adequate capacity to generalize over preferences. Extensive experiments show that our framework outperforms the best baselines by a large margin in terms of hypervolume in various real-world MOBO settings.