 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeChang-Yu Hsieh
Generative AI for Controllable Protein Sequence Design: A Survey
Feb 16, 2024
The design of novel protein sequences with targeted functionalities underpins a central theme in protein engineering, impacting diverse fields such as drug discovery and enzymatic engineering. However, navigating this vast combinatorial search space remains a severe challenge due to time and financial constraints. This scenario is rapidly evolving as the transformative advancements in AI, particularly in the realm of generative models and optimization algorithms, have been propelling the protein design field towards an unprecedented revolution. In this survey, we systematically review recent advances in generative AI for controllable protein sequence design. To set the stage, we first outline the foundational tasks in protein sequence design in terms of the constraints involved and present key generative models and optimization algorithms. We then offer in-depth reviews of each design task and discuss the pertinent applications. Finally, we identify the unresolved challenges and highlight research opportunities that merit deeper exploration.
From molecules to scaffolds to functional groups: building context-dependent molecular representation via multi-channel learning
Nov 05, 2023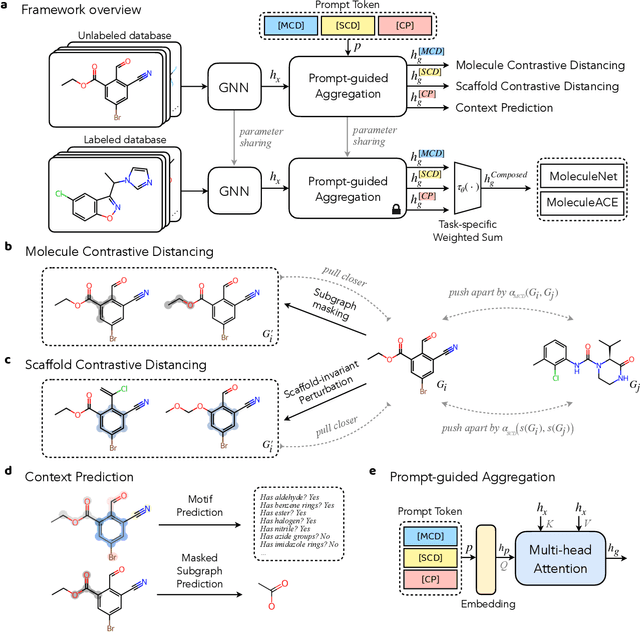
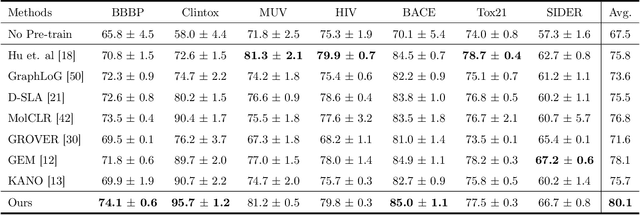
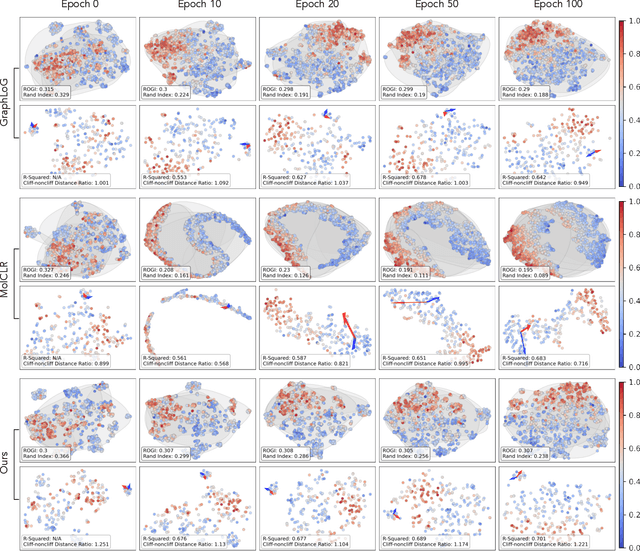
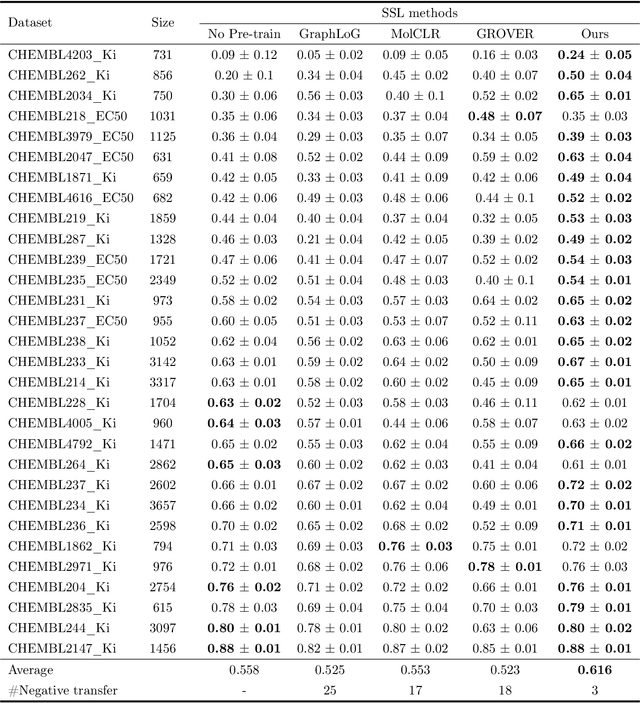
Reliable molecular property prediction is essential for various scientific endeavors and industrial applications, such as drug discovery. However, the scarcity of data, combined with the highly non-linear causal relationships between physicochemical and biological properties and conventional molecular featurization schemes, complicates the development of robust molecular machine learning models. Self-supervised learning (SSL) has emerged as a popular solution, utilizing large-scale, unannotated molecular data to learn a foundational representation of chemical space that might be advantageous for downstream tasks. Yet, existing molecular SSL methods largely overlook domain-specific knowledge, such as molecular similarity and scaffold importance, as well as the context of the target application when operating over the large chemical space. This paper introduces a novel learning framework that leverages the knowledge of structural hierarchies within molecular structures, embeds them through separate pre-training tasks over distinct channels, and employs a task-specific channel selection to compose a context-dependent representation. Our approach demonstrates competitive performance across various molecular property benchmarks and establishes some state-of-the-art results. It further offers unprecedented advantages in particularly challenging yet ubiquitous scenarios like activity cliffs with enhanced robustness and generalizability compared to other baselines.
MolHF: A Hierarchical Normalizing Flow for Molecular Graph Generation
May 15, 2023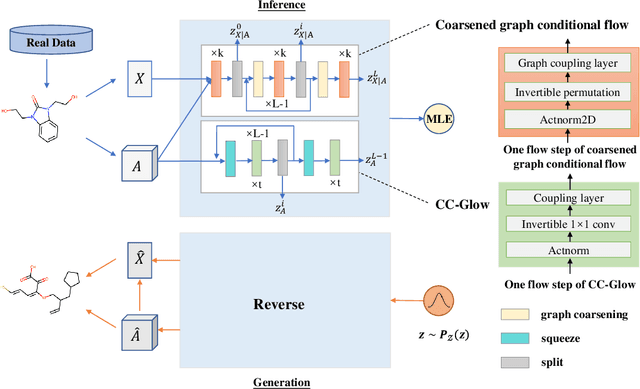
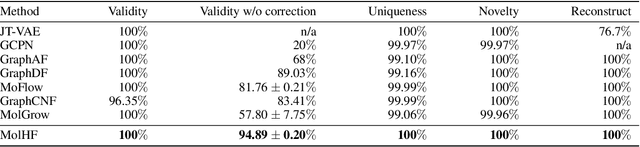
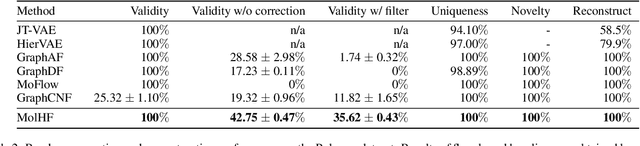
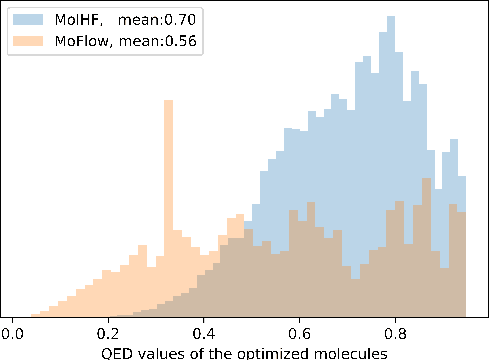
Molecular de novo design is a critical yet challenging task in scientific fields, aiming to design novel molecular structures with desired property profiles. Significant progress has been made by resorting to generative models for graphs. However, limited attention is paid to hierarchical generative models, which can exploit the inherent hierarchical structure (with rich semantic information) of the molecular graphs and generate complex molecules of larger size that we shall demonstrate to be difficult for most existing models. The primary challenge to hierarchical generation is the non-differentiable issue caused by the generation of intermediate discrete coarsened graph structures. To sidestep this issue, we cast the tricky hierarchical generation problem over discrete spaces as the reverse process of hierarchical representation learning and propose MolHF, a new hierarchical flow-based model that generates molecular graphs in a coarse-to-fine manner. Specifically, MolHF first generates bonds through a multi-scale architecture, then generates atoms based on the coarsened graph structure at each scale. We demonstrate that MolHF achieves state-of-the-art performance in random generation and property optimization, implying its high capacity to model data distribution. Furthermore, MolHF is the first flow-based model that can be applied to model larger molecules (polymer) with more than 100 heavy atoms. The code and models are available at https://github.com/violet-sto/MolHF.
An Equivariant Generative Framework for Molecular Graph-Structure Co-Design
Apr 12, 2023Designing molecules with desirable physiochemical properties and functionalities is a long-standing challenge in chemistry, material science, and drug discovery. Recently, machine learning-based generative models have emerged as promising approaches for \emph{de novo} molecule design. However, further refinement of methodology is highly desired as most existing methods lack unified modeling of 2D topology and 3D geometry information and fail to effectively learn the structure-property relationship for molecule design. Here we present MolCode, a roto-translation equivariant generative framework for \underline{Mol}ecular graph-structure \underline{Co-de}sign. In MolCode, 3D geometric information empowers the molecular 2D graph generation, which in turn helps guide the prediction of molecular 3D structure. Extensive experimental results show that MolCode outperforms previous methods on a series of challenging tasks including \emph{de novo} molecule design, targeted molecule discovery, and structure-based drug design. Particularly, MolCode not only consistently generates valid (99.95$\%$ Validity) and diverse (98.75$\%$ Uniqueness) molecular graphs/structures with desirable properties, but also generate drug-like molecules with high affinity to target proteins (61.8$\%$ high-affinity ratio), which demonstrates MolCode's potential applications in material design and drug discovery. Our extensive investigation reveals that the 2D topology and 3D geometry contain intrinsically complementary information in molecule design, and provide new insights into machine learning-based molecule representation and generation.
Sample-efficient Multi-objective Molecular Optimization with GFlowNets
Feb 08, 2023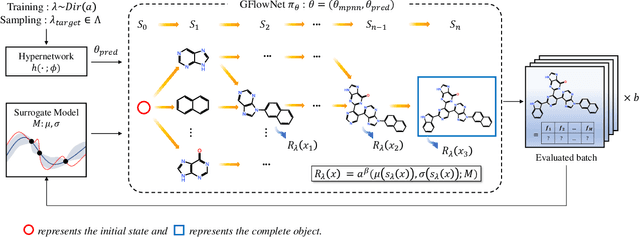
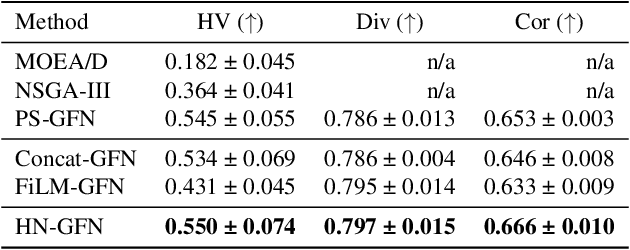
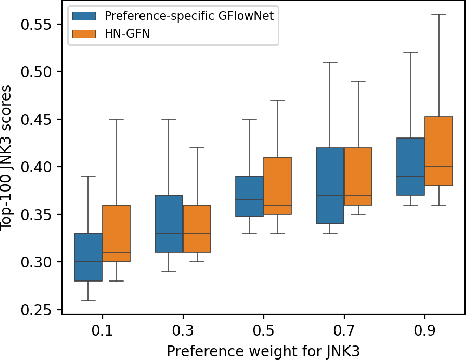
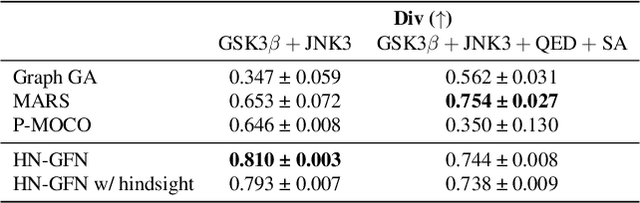
Many crucial scientific problems involve designing novel molecules with desired properties, which can be formulated as an expensive black-box optimization problem over the discrete chemical space. Computational methods have achieved initial success but still struggle with simultaneously optimizing multiple competing properties in a sample-efficient manner. In this work, we propose a multi-objective Bayesian optimization (MOBO) algorithm leveraging the hypernetwork-based GFlowNets (HN-GFN) as an acquisition function optimizer, with the purpose of sampling a diverse batch of candidate molecular graphs from an approximate Pareto front. Using a single preference-conditioned hypernetwork, HN-GFN learns to explore various trade-offs between objectives. Inspired by reinforcement learning, we further propose a hindsight-like off-policy strategy to share high-performing molecules among different preferences in order to speed up learning for HN-GFN. Through synthetic experiments, we illustrate that HN-GFN has adequate capacity to generalize over preferences. Extensive experiments show that our framework outperforms the best baselines by a large margin in terms of hypervolume in various real-world MOBO settings.
Protein-Ligand Complex Generator & Drug Screening via Tiered Tensor Transform
Jan 03, 2023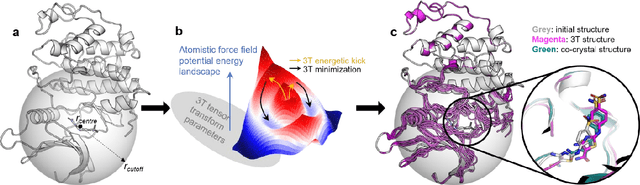
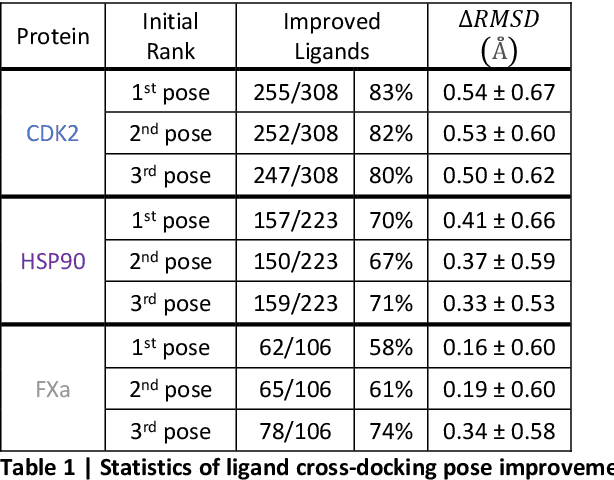
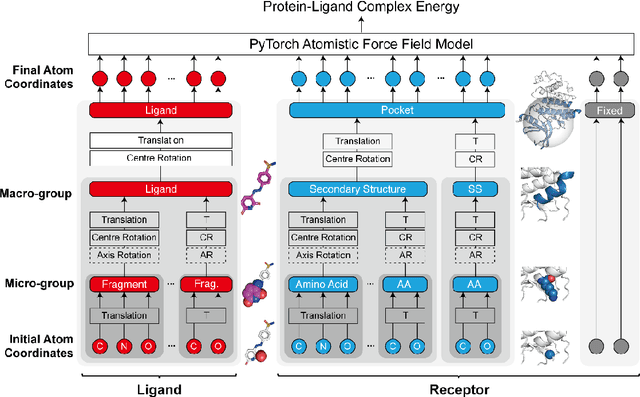
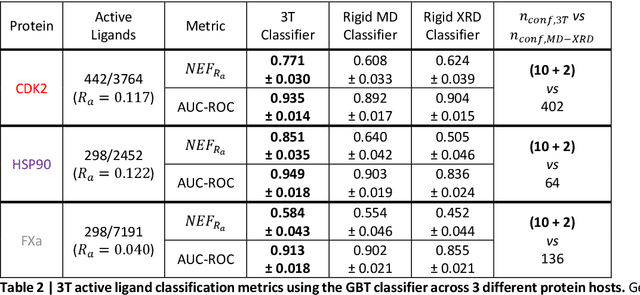
Accurate determination of a small molecule candidate (ligand) binding pose in its target protein pocket is important for computer-aided drug discovery. Typical rigid-body docking methods ignore the pocket flexibility of protein, while the more accurate pose generation using molecular dynamics is hindered by slow protein dynamics. We develop a tiered tensor transform (3T) algorithm to rapidly generate diverse protein-ligand complex conformations for both pose and affinity estimation in drug screening, requiring neither machine learning training nor lengthy dynamics computation, while maintaining both coarse-grain-like coordinated protein dynamics and atomistic-level details of the complex pocket. The 3T conformation structures we generate are closer to experimental co-crystal structures than those generated by docking software, and more importantly achieve significantly higher accuracy in active ligand classification than traditional ensemble docking using hundreds of experimental protein conformations. 3T structure transformation is decoupled from the system physics, making future usage in other computational scientific domains possible.
Retroformer: Pushing the Limits of Interpretable End-to-end Retrosynthesis Transformer
Jan 29, 2022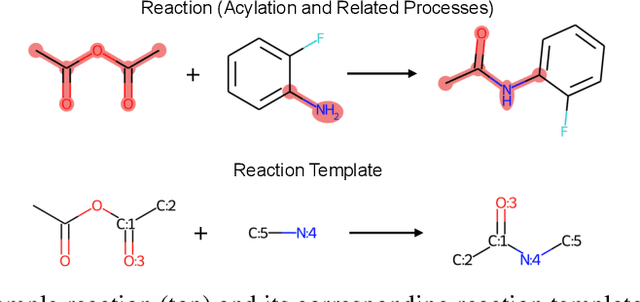
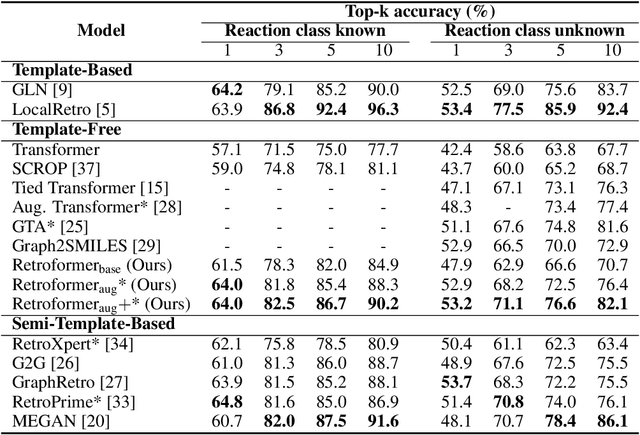
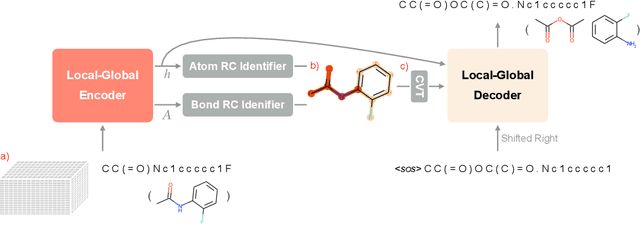
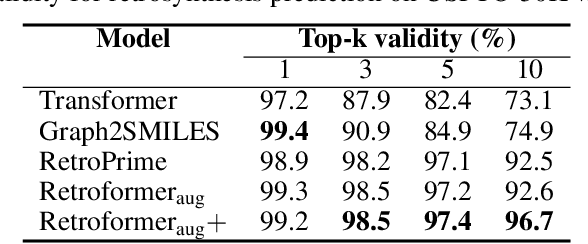
Retrosynthesis prediction is one of the fundamental challenges in organic synthesis. The task is to predict the reactants given a core product. With the advancement of machine learning, computer-aided synthesis planning has gained increasing interest. Numerous methods were proposed to solve this problem with different levels of dependency on additional chemical knowledge. In this paper, we propose Retroformer, a novel Transformer-based architecture for retrosynthesis prediction without relying on any cheminformatics tools for molecule editing. Via the proposed local attention head, the model can jointly encode the molecular sequence and graph, and efficiently exchange information between the local reactive region and the global reaction context. Retroformer reaches the new state-of-the-art accuracy for the end-to-end template-free retrosynthesis, and improves over many strong baselines on better molecule and reaction validity. In addition, its generative procedure is highly interpretable and controllable. Overall, Retroformer pushes the limits of the reaction reasoning ability of deep generative models.
Fast Extraction of Word Embedding from Q-contexts
Sep 15, 2021
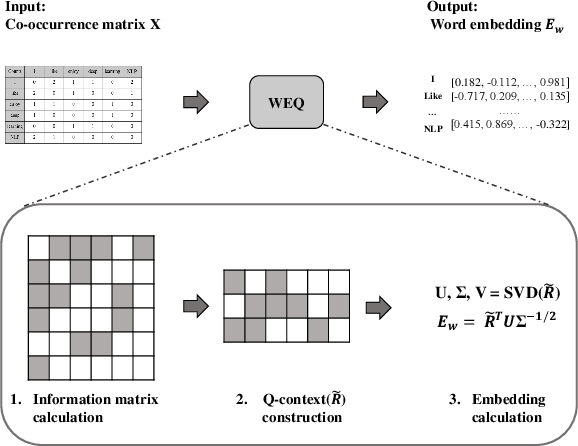
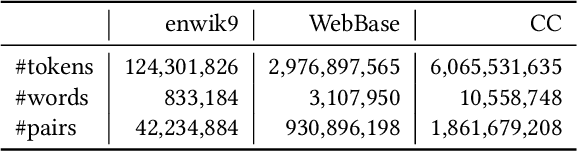
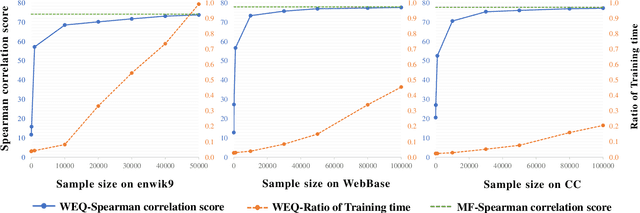
The notion of word embedding plays a fundamental role in natural language processing (NLP). However, pre-training word embedding for very large-scale vocabulary is computationally challenging for most existing methods. In this work, we show that with merely a small fraction of contexts (Q-contexts)which are typical in the whole corpus (and their mutual information with words), one can construct high-quality word embedding with negligible errors. Mutual information between contexts and words can be encoded canonically as a sampling state, thus, Q-contexts can be fast constructed. Furthermore, we present an efficient and effective WEQ method, which is capable of extracting word embedding directly from these typical contexts. In practical scenarios, our algorithm runs 11$\sim$13 times faster than well-established methods. By comparing with well-known methods such as matrix factorization, word2vec, GloVeand fasttext, we demonstrate that our method achieves comparable performance on a variety of downstream NLP tasks, and in the meanwhile maintains run-time and resource advantages over all these baselines.
Modeling Protein Using Large-scale Pretrain Language Model
Aug 17, 2021

Protein is linked to almost every life process. Therefore, analyzing the biological structure and property of protein sequences is critical to the exploration of life, as well as disease detection and drug discovery. Traditional protein analysis methods tend to be labor-intensive and time-consuming. The emergence of deep learning models makes modeling data patterns in large quantities of data possible. Interdisciplinary researchers have begun to leverage deep learning methods to model large biological datasets, e.g. using long short-term memory and convolutional neural network for protein sequence classification. After millions of years of evolution, evolutionary information is encoded in protein sequences. Inspired by the similarity between natural language and protein sequences, we use large-scale language models to model evolutionary-scale protein sequences, encoding protein biology information in representation. Significant improvements are observed in both token-level and sequence-level tasks, demonstrating that our large-scale model can accurately capture evolution information from pretraining on evolutionary-scale individual sequences. Our code and model are available at https://github.com/THUDM/ProteinLM.
Quantum algorithm for training nonlinear SVMs in almost linear time
Jun 18, 2020
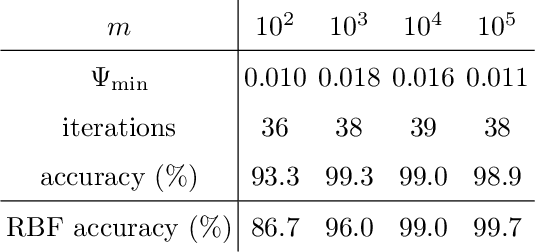
We propose a quantum algorithm for training nonlinear support vector machines (SVM) for feature space learning where classical input data is encoded in the amplitudes of quantum states. Based on the classical algorithm of Joachims, our algorithm has a running time which scales linearly in the number of training examples (up to polylogarithmic factors) and applies to the standard soft-margin $\ell_1$-SVM model. In contrast, the best classical algorithms have super-linear scaling for general feature maps, and achieve linear $m$ scaling only for linear SVMs, where classification is performed in the original input data space, or for the special case of feature maps corresponding to shift-invariant kernels. Similarly, previously proposed quantum algorithms either have super-linear scaling in $m$, or else apply to different SVM models such as the hard-margin or least squares $\ell_2$-SVM, which lack certain desirable properties of the soft-margin $\ell_1$-SVM model. We classically simulate our algorithm and give evidence that it can perform well in practice, and not only for asymptotically large data sets.