 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeZaixi Zhang
FedGT: Federated Node Classification with Scalable Graph Transformer
Jan 26, 2024
Graphs are widely used to model relational data. As graphs are getting larger and larger in real-world scenarios, there is a trend to store and compute subgraphs in multiple local systems. For example, recently proposed \emph{subgraph federated learning} methods train Graph Neural Networks (GNNs) distributively on local subgraphs and aggregate GNN parameters with a central server. However, existing methods have the following limitations: (1) The links between local subgraphs are missing in subgraph federated learning. This could severely damage the performance of GNNs that follow message-passing paradigms to update node/edge features. (2) Most existing methods overlook the subgraph heterogeneity issue, brought by subgraphs being from different parts of the whole graph. To address the aforementioned challenges, we propose a scalable \textbf{Fed}erated \textbf{G}raph \textbf{T}ransformer (\textbf{FedGT}) in the paper. Firstly, we design a hybrid attention scheme to reduce the complexity of the Graph Transformer to linear while ensuring a global receptive field with theoretical bounds. Specifically, each node attends to the sampled local neighbors and a set of curated global nodes to learn both local and global information and be robust to missing links. The global nodes are dynamically updated during training with an online clustering algorithm to capture the data distribution of the corresponding local subgraph. Secondly, FedGT computes clients' similarity based on the aligned global nodes with optimal transport. The similarity is then used to perform weighted averaging for personalized aggregation, which well addresses the data heterogeneity problem. Moreover, local differential privacy is applied to further protect the privacy of clients. Finally, extensive experimental results on 6 datasets and 2 subgraph settings demonstrate the superiority of FedGT.
Multi-scale Iterative Refinement towards Robust and Versatile Molecular Docking
Nov 30, 2023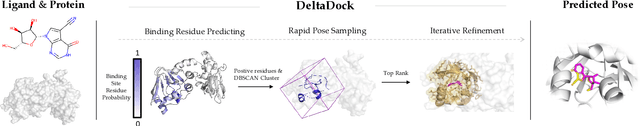
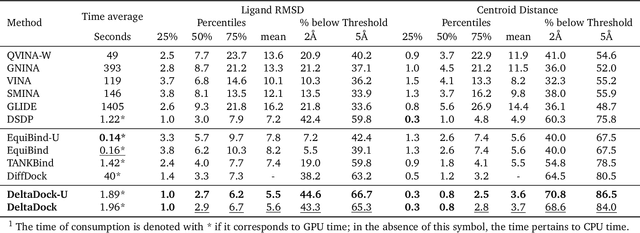
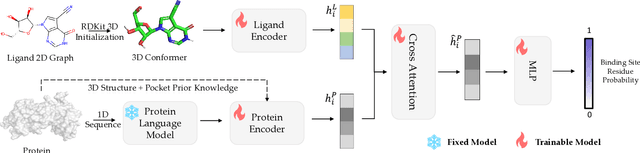
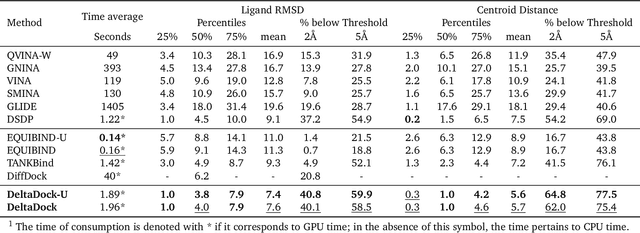
Molecular docking is a key computational tool utilized to predict the binding conformations of small molecules to protein targets, which is fundamental in the design of novel drugs. Despite recent advancements in geometric deep learning-based approaches leading to improvements in blind docking efficiency, these methods have encountered notable challenges, such as limited generalization performance on unseen proteins, the inability to concurrently address the settings of blind docking and site-specific docking, and the frequent occurrence of physical implausibilities such as inter-molecular steric clash. In this study, we introduce DeltaDock, a robust and versatile framework designed for efficient molecular docking to overcome these challenges. DeltaDock operates in a two-step process: rapid initial complex structures sampling followed by multi-scale iterative refinement of the initial structures. In the initial stage, to sample accurate structures with high efficiency, we develop a ligand-dependent binding site prediction model founded on large protein models and graph neural networks. This model is then paired with GPU-accelerated sampling algorithms. The sampled structures are updated using a multi-scale iterative refinement module that captures both protein-ligand atom-atom interactions and residue-atom interactions in the following stage. Distinct from previous geometric deep learning methods that are conditioned on the blind docking setting, DeltaDock demonstrates superior performance in both blind docking and site-specific docking settings. Comprehensive experimental results reveal that DeltaDock consistently surpasses baseline methods in terms of docking accuracy. Furthermore, it displays remarkable generalization capabilities and proficiency for predicting physically valid structures, thereby attesting to its robustness and reliability in various scenarios.
Sparse Attention-Based Neural Networks for Code Classification
Nov 11, 2023Categorizing source codes accurately and efficiently is a challenging problem in real-world programming education platform management. In recent years, model-based approaches utilizing abstract syntax trees (ASTs) have been widely applied to code classification tasks. We introduce an approach named the Sparse Attention-based neural network for Code Classification (SACC) in this paper. The approach involves two main steps: In the first step, source code undergoes syntax parsing and preprocessing. The generated abstract syntax tree is split into sequences of subtrees and then encoded using a recursive neural network to obtain a high-dimensional representation. This step simultaneously considers both the logical structure and lexical level information contained within the code. In the second step, the encoded sequences of subtrees are fed into a Transformer model that incorporates sparse attention mechanisms for the purpose of classification. This method efficiently reduces the computational cost of the self-attention mechanisms, thus improving the training speed while preserving effectiveness. Our work introduces a carefully designed sparse attention pattern that is specifically designed to meet the unique needs of code classification tasks. This design helps reduce the influence of redundant information and enhances the overall performance of the model. Finally, we also deal with problems in previous related research, which include issues like incomplete classification labels and a small dataset size. We annotated the CodeNet dataset with algorithm-related labeling categories, which contains a significantly large amount of data. Extensive comparative experimental results demonstrate the effectiveness and efficiency of SACC for the code classification tasks.
AdaptSSR: Pre-training User Model with Augmentation-Adaptive Self-Supervised Ranking
Oct 24, 2023



User modeling, which aims to capture users' characteristics or interests, heavily relies on task-specific labeled data and suffers from the data sparsity issue. Several recent studies tackled this problem by pre-training the user model on massive user behavior sequences with a contrastive learning task. Generally, these methods assume different views of the same behavior sequence constructed via data augmentation are semantically consistent, i.e., reflecting similar characteristics or interests of the user, and thus maximizing their agreement in the feature space. However, due to the diverse interests and heavy noise in user behaviors, existing augmentation methods tend to lose certain characteristics of the user or introduce noisy behaviors. Thus, forcing the user model to directly maximize the similarity between the augmented views may result in a negative transfer. To this end, we propose to replace the contrastive learning task with a new pretext task: Augmentation-Adaptive SelfSupervised Ranking (AdaptSSR), which alleviates the requirement of semantic consistency between the augmented views while pre-training a discriminative user model. Specifically, we adopt a multiple pairwise ranking loss which trains the user model to capture the similarity orders between the implicitly augmented view, the explicitly augmented view, and views from other users. We further employ an in-batch hard negative sampling strategy to facilitate model training. Moreover, considering the distinct impacts of data augmentation on different behavior sequences, we design an augmentation-adaptive fusion mechanism to automatically adjust the similarity order constraint applied to each sample based on the estimated similarity between the augmented views. Extensive experiments on both public and industrial datasets with six downstream tasks verify the effectiveness of AdaptSSR.
A Systematic Survey in Geometric Deep Learning for Structure-based Drug Design
Jul 03, 2023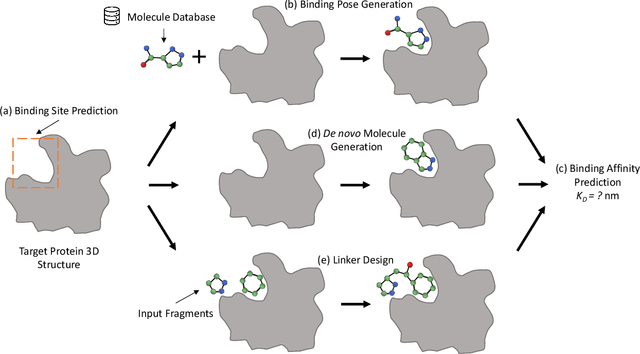

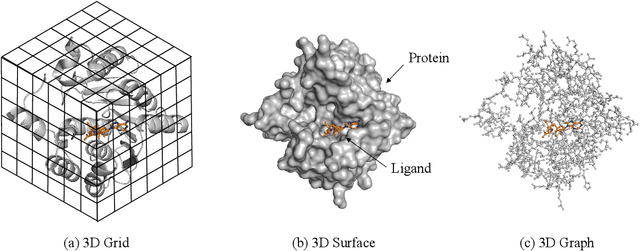

Structure-based drug design (SBDD), which utilizes the three-dimensional geometry of proteins to identify potential drug candidates, is becoming increasingly vital in drug discovery. However, traditional methods based on physiochemical modeling and experts' domain knowledge are time-consuming and laborious. The recent advancements in geometric deep learning, which integrates and processes 3D geometric data, coupled with the availability of accurate protein 3D structure predictions from tools like AlphaFold, have significantly propelled progress in structure-based drug design. In this paper, we systematically review the recent progress of geometric deep learning for structure-based drug design. We start with a brief discussion of the mainstream tasks in structure-based drug design, commonly used 3D protein representations and representative predictive/generative models. Then we delve into detailed reviews for each task (binding site prediction, binding pose generation, \emph{de novo} molecule generation, linker design, and binding affinity prediction), including the problem setup, representative methods, datasets, and evaluation metrics. Finally, we conclude this survey with the current challenges and highlight potential opportunities of geometric deep learning for structure-based drug design.We curate a GitHub repo containing the related papers \url{https://github.com/zaixizhang/Awesome-SBDD}.
Learning Subpocket Prototypes for Generalizable Structure-based Drug Design
May 22, 2023



Generating molecules with high binding affinities to target proteins (a.k.a. structure-based drug design) is a fundamental and challenging task in drug discovery. Recently, deep generative models have achieved remarkable success in generating 3D molecules conditioned on the protein pocket. However, most existing methods consider molecular generation for protein pockets independently while neglecting the underlying connections such as subpocket-level similarities. Subpockets are the local protein environments of ligand fragments and pockets with similar subpockets may bind the same molecular fragment (motif) even though their overall structures are different. Therefore, the trained models can hardly generalize to unseen protein pockets in real-world applications. In this paper, we propose a novel method DrugGPS for generalizable structure-based drug design. With the biochemical priors, we propose to learn subpocket prototypes and construct a global interaction graph to model the interactions between subpocket prototypes and molecular motifs. Moreover, a hierarchical graph transformer encoder and motif-based 3D molecule generation scheme are used to improve the model's performance. The experimental results show that our model consistently outperforms baselines in generating realistic drug candidates with high affinities in challenging out-of-distribution settings.
An Equivariant Generative Framework for Molecular Graph-Structure Co-Design
Apr 12, 2023Designing molecules with desirable physiochemical properties and functionalities is a long-standing challenge in chemistry, material science, and drug discovery. Recently, machine learning-based generative models have emerged as promising approaches for \emph{de novo} molecule design. However, further refinement of methodology is highly desired as most existing methods lack unified modeling of 2D topology and 3D geometry information and fail to effectively learn the structure-property relationship for molecule design. Here we present MolCode, a roto-translation equivariant generative framework for \underline{Mol}ecular graph-structure \underline{Co-de}sign. In MolCode, 3D geometric information empowers the molecular 2D graph generation, which in turn helps guide the prediction of molecular 3D structure. Extensive experimental results show that MolCode outperforms previous methods on a series of challenging tasks including \emph{de novo} molecule design, targeted molecule discovery, and structure-based drug design. Particularly, MolCode not only consistently generates valid (99.95$\%$ Validity) and diverse (98.75$\%$ Uniqueness) molecular graphs/structures with desirable properties, but also generate drug-like molecules with high affinity to target proteins (61.8$\%$ high-affinity ratio), which demonstrates MolCode's potential applications in material design and drug discovery. Our extensive investigation reveals that the 2D topology and 3D geometry contain intrinsically complementary information in molecule design, and provide new insights into machine learning-based molecule representation and generation.
Backdoor Defense via Deconfounded Representation Learning
Mar 13, 2023



Deep neural networks (DNNs) are recently shown to be vulnerable to backdoor attacks, where attackers embed hidden backdoors in the DNN model by injecting a few poisoned examples into the training dataset. While extensive efforts have been made to detect and remove backdoors from backdoored DNNs, it is still not clear whether a backdoor-free clean model can be directly obtained from poisoned datasets. In this paper, we first construct a causal graph to model the generation process of poisoned data and find that the backdoor attack acts as the confounder, which brings spurious associations between the input images and target labels, making the model predictions less reliable. Inspired by the causal understanding, we propose the Causality-inspired Backdoor Defense (CBD), to learn deconfounded representations for reliable classification. Specifically, a backdoored model is intentionally trained to capture the confounding effects. The other clean model dedicates to capturing the desired causal effects by minimizing the mutual information with the confounding representations from the backdoored model and employing a sample-wise re-weighting scheme. Extensive experiments on multiple benchmark datasets against 6 state-of-the-art attacks verify that our proposed defense method is effective in reducing backdoor threats while maintaining high accuracy in predicting benign samples. Further analysis shows that CBD can also resist potential adaptive attacks. The code is available at \url{https://github.com/zaixizhang/CBD}.
Untargeted Attack against Federated Recommendation Systems via Poisonous Item Embeddings and the Defense
Dec 11, 2022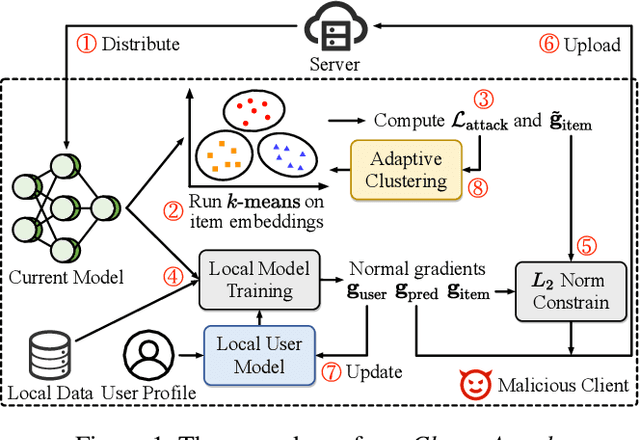
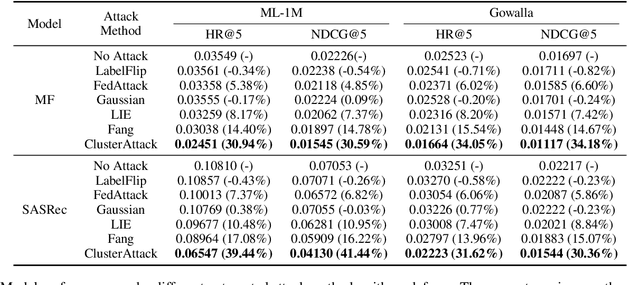

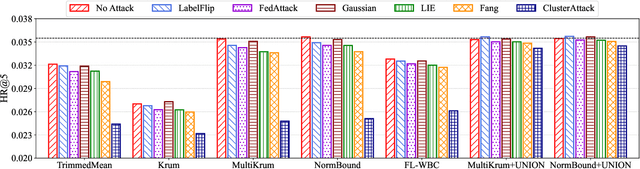
Federated recommendation (FedRec) can train personalized recommenders without collecting user data, but the decentralized nature makes it susceptible to poisoning attacks. Most previous studies focus on the targeted attack to promote certain items, while the untargeted attack that aims to degrade the overall performance of the FedRec system remains less explored. In fact, untargeted attacks can disrupt the user experience and bring severe financial loss to the service provider. However, existing untargeted attack methods are either inapplicable or ineffective against FedRec systems. In this paper, we delve into the untargeted attack and its defense for FedRec systems. (i) We propose ClusterAttack, a novel untargeted attack method. It uploads poisonous gradients that converge the item embeddings into several dense clusters, which make the recommender generate similar scores for these items in the same cluster and perturb the ranking order. (ii) We propose a uniformity-based defense mechanism (UNION) to protect FedRec systems from such attacks. We design a contrastive learning task that regularizes the item embeddings toward a uniform distribution. Then the server filters out these malicious gradients by estimating the uniformity of updated item embeddings. Experiments on two public datasets show that ClusterAttack can effectively degrade the performance of FedRec systems while circumventing many defense methods, and UNION can improve the resistance of the system against various untargeted attacks, including our ClusterAttack.
Hierarchical Graph Transformer with Adaptive Node Sampling
Oct 08, 2022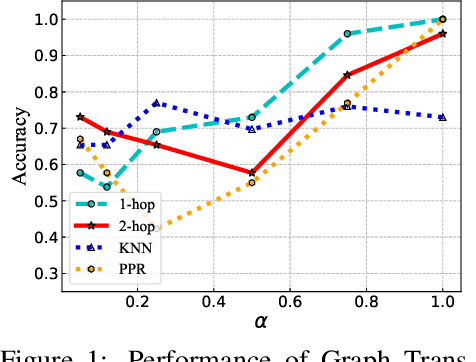
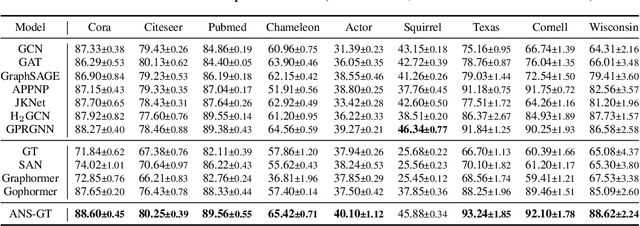
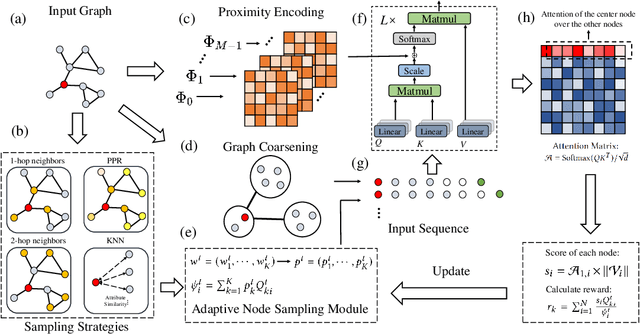
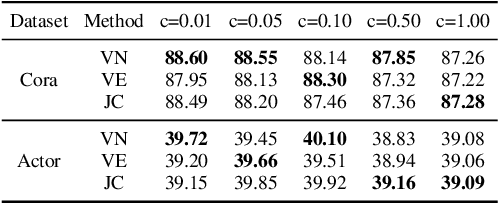
The Transformer architecture has achieved remarkable success in a number of domains including natural language processing and computer vision. However, when it comes to graph-structured data, transformers have not achieved competitive performance, especially on large graphs. In this paper, we identify the main deficiencies of current graph transformers:(1) Existing node sampling strategies in Graph Transformers are agnostic to the graph characteristics and the training process. (2) Most sampling strategies only focus on local neighbors and neglect the long-range dependencies in the graph. We conduct experimental investigations on synthetic datasets to show that existing sampling strategies are sub-optimal. To tackle the aforementioned problems, we formulate the optimization strategies of node sampling in Graph Transformer as an adversary bandit problem, where the rewards are related to the attention weights and can vary in the training procedure. Meanwhile, we propose a hierarchical attention scheme with graph coarsening to capture the long-range interactions while reducing computational complexity. Finally, we conduct extensive experiments on real-world datasets to demonstrate the superiority of our method over existing graph transformers and popular GNNs.