 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeKirill Neklyudov
Diffusion Models as Constrained Samplers for Optimization with Unknown Constraints
Feb 28, 2024
Addressing real-world optimization problems becomes particularly challenging when analytic objective functions or constraints are unavailable. While numerous studies have addressed the issue of unknown objectives, limited research has focused on scenarios where feasibility constraints are not given explicitly. Overlooking these constraints can lead to spurious solutions that are unrealistic in practice. To deal with such unknown constraints, we propose to perform optimization within the data manifold using diffusion models. To constrain the optimization process to the data manifold, we reformulate the original optimization problem as a sampling problem from the product of the Boltzmann distribution defined by the objective function and the data distribution learned by the diffusion model. To enhance sampling efficiency, we propose a two-stage framework that begins with a guided diffusion process for warm-up, followed by a Langevin dynamics stage for further correction. Theoretical analysis shows that the initial stage results in a distribution focused on feasible solutions, thereby providing a better initialization for the later stage. Comprehensive experiments on a synthetic dataset, six real-world black-box optimization datasets, and a multi-objective optimization dataset show that our method achieves better or comparable performance with previous state-of-the-art baselines.
Structured Inverse-Free Natural Gradient: Memory-Efficient & Numerically-Stable KFAC for Large Neural Nets
Dec 16, 2023Second-order methods for deep learning -- such as KFAC -- can be useful for neural net training. However, they are often memory-inefficient and numerically unstable for low-precision training since their preconditioning Kronecker factors are dense, and require high-precision matrix inversion or decomposition. Consequently, such methods are not widely used for training large neural networks such as transformer-based models. We address these two issues by (i) formulating an inverse-free update of KFAC and (ii) imposing structures in each of the Kronecker factors, resulting in a method we term structured inverse-free natural gradient descent (SINGD). On large modern neural networks, we show that, in contrast to KFAC, SINGD is memory efficient and numerically robust, and often outperforms AdamW even in half precision. Hence, our work closes a gap between first-order and second-order methods in modern low precision training for large neural nets.
A Computational Framework for Solving Wasserstein Lagrangian Flows
Oct 17, 2023The dynamical formulation of the optimal transport can be extended through various choices of the underlying geometry ($\textit{kinetic energy}$), and the regularization of density paths ($\textit{potential energy}$). These combinations yield different variational problems ($\textit{Lagrangians}$), encompassing many variations of the optimal transport problem such as the Schr\"odinger bridge, unbalanced optimal transport, and optimal transport with physical constraints, among others. In general, the optimal density path is unknown, and solving these variational problems can be computationally challenging. Leveraging the dual formulation of the Lagrangians, we propose a novel deep learning based framework approaching all of these problems from a unified perspective. Our method does not require simulating or backpropagating through the trajectories of the learned dynamics, and does not need access to optimal couplings. We showcase the versatility of the proposed framework by outperforming previous approaches for the single-cell trajectory inference, where incorporating prior knowledge into the dynamics is crucial for correct predictions.
Wasserstein Quantum Monte Carlo: A Novel Approach for Solving the Quantum Many-Body Schrödinger Equation
Jul 17, 2023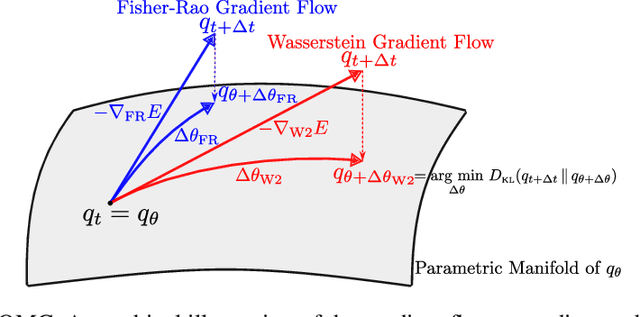

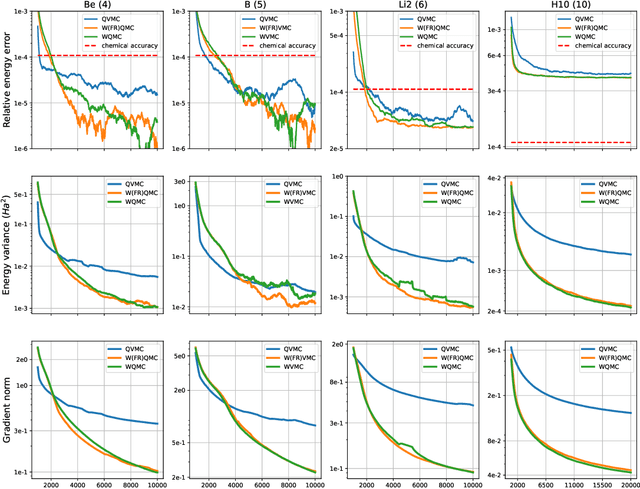
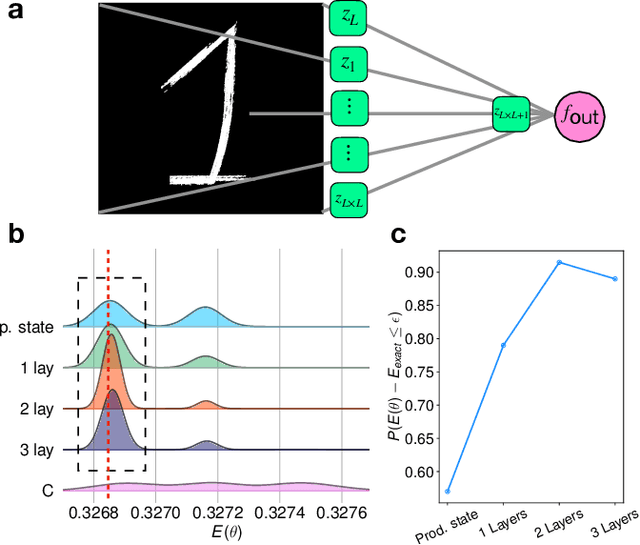
Solving the quantum many-body Schr\"odinger equation is a fundamental and challenging problem in the fields of quantum physics, quantum chemistry, and material sciences. One of the common computational approaches to this problem is Quantum Variational Monte Carlo (QVMC), in which ground-state solutions are obtained by minimizing the energy of the system within a restricted family of parameterized wave functions. Deep learning methods partially address the limitations of traditional QVMC by representing a rich family of wave functions in terms of neural networks. However, the optimization objective in QVMC remains notoriously hard to minimize and requires second-order optimization methods such as natural gradient. In this paper, we first reformulate energy functional minimization in the space of Born distributions corresponding to particle-permutation (anti-)symmetric wave functions, rather than the space of wave functions. We then interpret QVMC as the Fisher-Rao gradient flow in this distributional space, followed by a projection step onto the variational manifold. This perspective provides us with a principled framework to derive new QMC algorithms, by endowing the distributional space with better metrics, and following the projected gradient flow induced by those metrics. More specifically, we propose "Wasserstein Quantum Monte Carlo" (WQMC), which uses the gradient flow induced by the Wasserstein metric, rather than Fisher-Rao metric, and corresponds to transporting the probability mass, rather than teleporting it. We demonstrate empirically that the dynamics of WQMC results in faster convergence to the ground state of molecular systems.
Quantum HyperNetworks: Training Binary Neural Networks in Quantum Superposition
Jan 19, 2023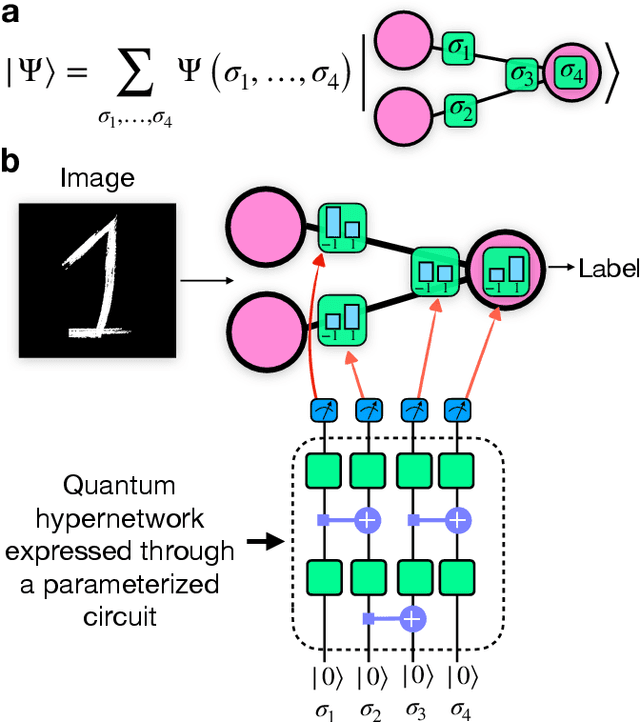
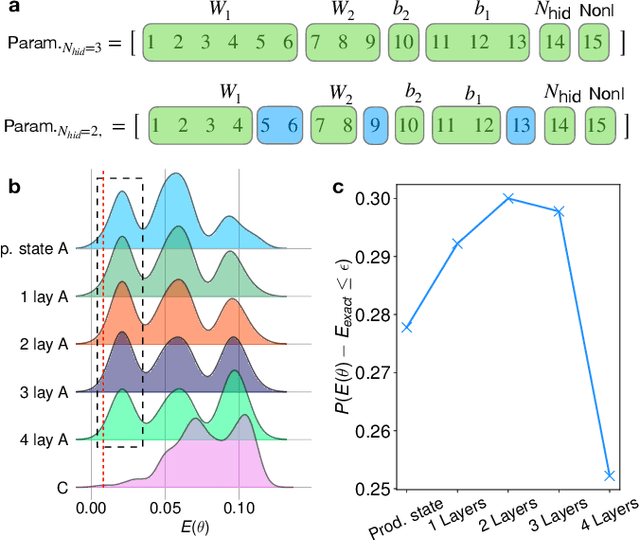
Binary neural networks, i.e., neural networks whose parameters and activations are constrained to only two possible values, offer a compelling avenue for the deployment of deep learning models on energy- and memory-limited devices. However, their training, architectural design, and hyperparameter tuning remain challenging as these involve multiple computationally expensive combinatorial optimization problems. Here we introduce quantum hypernetworks as a mechanism to train binary neural networks on quantum computers, which unify the search over parameters, hyperparameters, and architectures in a single optimization loop. Through classical simulations, we demonstrate that of our approach effectively finds optimal parameters, hyperparameters and architectural choices with high probability on classification problems including a two-dimensional Gaussian dataset and a scaled-down version of the MNIST handwritten digits. We represent our quantum hypernetworks as variational quantum circuits, and find that an optimal circuit depth maximizes the probability of finding performant binary neural networks. Our unified approach provides an immense scope for other applications in the field of machine learning.
Action Matching: A Variational Method for Learning Stochastic Dynamics from Samples
Oct 13, 2022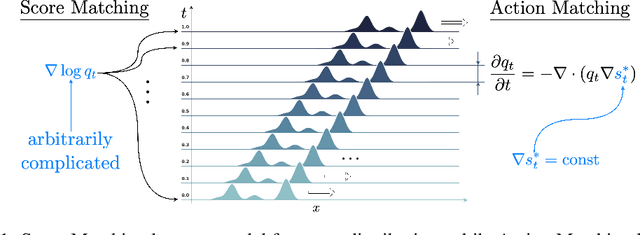
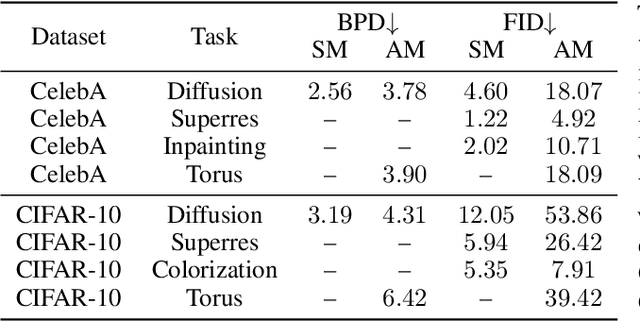
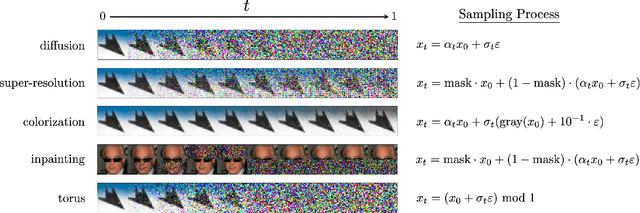
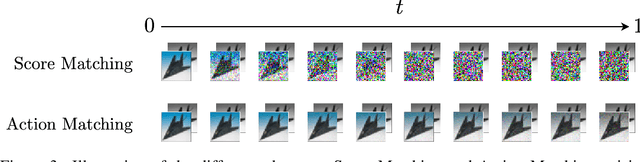
Stochastic dynamics are ubiquitous in many fields of science, from the evolution of quantum systems in physics to diffusion-based models in machine learning. Existing methods such as score matching can be used to simulate these physical processes by assuming that the dynamics is a diffusion, which is not always the case. In this work, we propose a method called "Action Matching" that enables us to learn a much broader family of stochastic dynamics. Our method requires access only to samples from different time-steps, makes no explicit assumptions about the underlying dynamics, and can be applied even when samples are uncorrelated (i.e., are not part of a trajectory). Action Matching directly learns an underlying mechanism to move samples in time without modeling the distributions at each time-step. In this work, we showcase how Action Matching can be used for several computer vision tasks such as generative modeling, super-resolution, colorization, and inpainting; and further discuss potential applications in other areas of science.
Particle Dynamics for Learning EBMs
Nov 26, 2021

Energy-based modeling is a promising approach to unsupervised learning, which yields many downstream applications from a single model. The main difficulty in learning energy-based models with the "contrastive approaches" is the generation of samples from the current energy function at each iteration. Many advances have been made to accomplish this subroutine cheaply. Nevertheless, all such sampling paradigms run MCMC targeting the current model, which requires infinitely long chains to generate samples from the true energy distribution and is problematic in practice. This paper proposes an alternative approach to getting these samples and avoiding crude MCMC sampling from the current model. We accomplish this by viewing the evolution of the modeling distribution as (i) the evolution of the energy function, and (ii) the evolution of the samples from this distribution along some vector field. We subsequently derive this time-dependent vector field such that the particles following this field are approximately distributed as the current density model. Thereby we match the evolution of the particles with the evolution of the energy function prescribed by the learning procedure. Importantly, unlike Monte Carlo sampling, our method targets to match the current distribution in a finite time. Finally, we demonstrate its effectiveness empirically compared to MCMC-based learning methods.
Deterministic Gibbs Sampling via Ordinary Differential Equations
Jun 18, 2021



Deterministic dynamics is an essential part of many MCMC algorithms, e.g. Hybrid Monte Carlo or samplers utilizing normalizing flows. This paper presents a general construction of deterministic measure-preserving dynamics using autonomous ODEs and tools from differential geometry. We show how Hybrid Monte Carlo and other deterministic samplers follow as special cases of our theory. We then demonstrate the utility of our approach by constructing a continuous non-sequential version of Gibbs sampling in terms of an ODE flow and extending it to discrete state spaces. We find that our deterministic samplers are more sample efficient than stochastic counterparts, even if the latter generate independent samples.
Orbital MCMC
Oct 15, 2020



Markov Chain Monte Carlo (MCMC) is a computational approach to fundamental problems such as inference, integration, optimization, and simulation. Recently, the framework (Involutive MCMC) was proposed describing a large body of MCMC algorithms via two components: a stochastic acceptance test and an involutive deterministic function. This paper demonstrates that this framework is a special case of a larger family of algorithms operating on orbits of continuous deterministic bijections. We describe this family by deriving a novel MCMC kernel, which we call orbital MCMC (oMCMC). We provide a theoretical analysis and illustrate its utility using simple examples.
Involutive MCMC: a Unifying Framework
Jun 30, 2020


Markov Chain Monte Carlo (MCMC) is a computational approach to fundamental problems such as inference, integration, optimization, and simulation. The field has developed a broad spectrum of algorithms, varying in the way they are motivated, the way they are applied and how efficiently they sample. Despite all the differences, many of them share the same core principle, which we unify as the Involutive MCMC (iMCMC) framework. Building upon this, we describe a wide range of MCMC algorithms in terms of iMCMC, and formulate a number of "tricks" which one can use as design principles for developing new MCMC algorithms. Thus, iMCMC provides a unified view of many known MCMC algorithms, which facilitates the derivation of powerful extensions. We demonstrate the latter with two examples where we transform known reversible MCMC algorithms into more efficient irreversible ones.