 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMao Su
Physical formula enhanced multi-task learning for pharmacokinetics prediction
Apr 16, 2024
Artificial intelligence (AI) technology has demonstrated remarkable potential in drug dis-covery, where pharmacokinetics plays a crucial role in determining the dosage, safety, and efficacy of new drugs. A major challenge for AI-driven drug discovery (AIDD) is the scarcity of high-quality data, which often requires extensive wet-lab work. A typical example of this is pharmacokinetic experiments. In this work, we develop a physical formula enhanced mul-ti-task learning (PEMAL) method that predicts four key parameters of pharmacokinetics simultaneously. By incorporating physical formulas into the multi-task framework, PEMAL facilitates effective knowledge sharing and target alignment among the pharmacokinetic parameters, thereby enhancing the accuracy of prediction. Our experiments reveal that PEMAL significantly lowers the data demand, compared to typical Graph Neural Networks. Moreover, we demonstrate that PEMAL enhances the robustness to noise, an advantage that conventional Neural Networks do not possess. Another advantage of PEMAL is its high flexibility, which can be potentially applied to other multi-task machine learning scenarios. Overall, our work illustrates the benefits and potential of using PEMAL in AIDD and other scenarios with data scarcity and noise.
ChemLLM: A Chemical Large Language Model
Feb 10, 2024Large language models (LLMs) have made impressive progress in chemistry applications, including molecular property prediction, molecular generation, experimental protocol design, etc. However, the community lacks a dialogue-based model specifically designed for chemistry. The challenge arises from the fact that most chemical data and scientific knowledge are primarily stored in structured databases, and the direct use of these structured data compromises the model's ability to maintain coherent dialogue. To tackle this issue, we develop a novel template-based instruction construction method that transforms structured knowledge into plain dialogue, making it suitable for language model training. By leveraging this approach, we develop ChemLLM, the first large language model dedicated to chemistry, capable of performing various tasks across chemical disciplines with smooth dialogue interaction. ChemLLM beats GPT-3.5 on all three principal tasks in chemistry, i.e., name conversion, molecular caption, and reaction prediction, and surpasses GPT-4 on two of them. Remarkably, ChemLLM also shows exceptional adaptability to related mathematical and physical tasks despite being trained mainly on chemical-centric corpora. Furthermore, ChemLLM demonstrates proficiency in specialized NLP tasks within chemistry, such as literature translation and cheminformatic programming. ChemLLM opens up a new avenue for exploration within chemical studies, while our method of integrating structured chemical knowledge into dialogue systems sets a new frontier for developing LLMs across various scientific fields. Codes, Datasets, and Model weights are publicly accessible at hf.co/AI4Chem/ChemLLM-7B-Chat.
Transferable E(3) equivariant parameterization for Hamiltonian of molecules and solids
Oct 28, 2022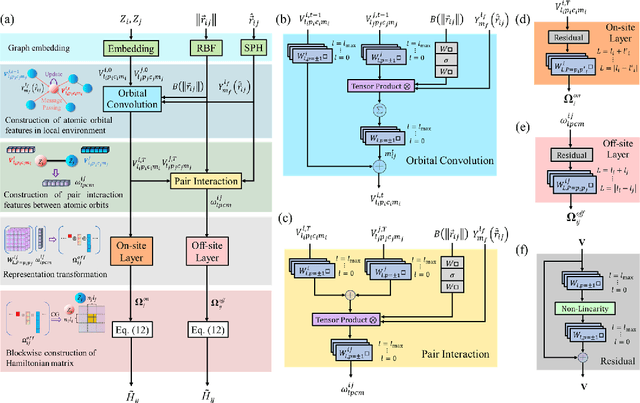

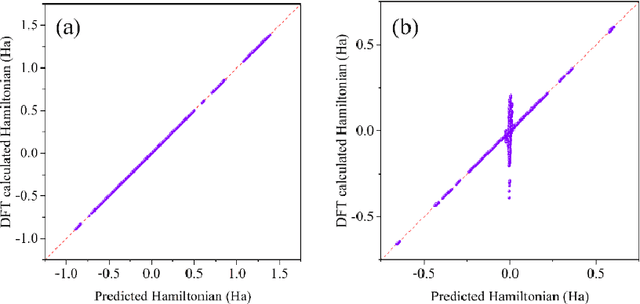
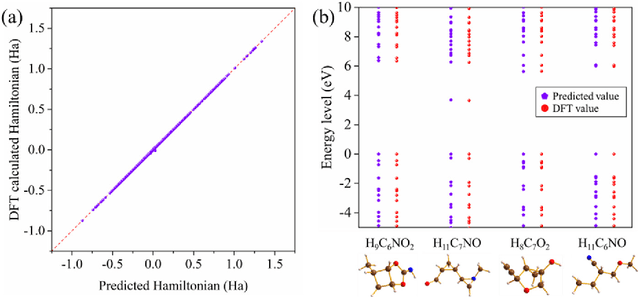
Machine learning, especially deep learning, can build a direct mapping from structure to properties with its huge parameter space, making it possible to perform high-throughput screening for the desired properties of materials. However, since the electronic Hamiltonian transforms non-trivially under rotation operations, it is challenging to accurately predict the electronic Hamiltonian while strictly satisfying this constraint. There is currently a lack of transferable machine learning models that can bypass the computationally demanding density functional theory (DFT) to obtain the ab initio Hamiltonian of molecules and materials by complete data-driven methods. In this work, we point out the necessity of explicitly considering the parity symmetry of the electronic Hamiltonian in addition to rotational equivariance. We propose a parameterized Hamiltonian that strictly satisfies rotational equivariance and parity symmetry simultaneously, based on which we develop an E(3) equivariant neural network called HamNet to predict the ab initio tight-binding Hamiltonian of various molecules and solids. The tests show that this model has similar transferability to that of machine learning potentials and can be applied to a class of materials with different configurations using the same set of trained network weights. The proposed framework provides a general transferable model for accelerating electronic structure calculations.