 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeShahira Abousamra
TopoSemiSeg: Enforcing Topological Consistency for Semi-Supervised Segmentation of Histopathology Images
Dec 06, 2023
In computational pathology, segmenting densely distributed objects like glands and nuclei is crucial for downstream analysis. To alleviate the burden of obtaining pixel-wise annotations, semi-supervised learning methods learn from large amounts of unlabeled data. Nevertheless, existing semi-supervised methods overlook the topological information hidden in the unlabeled images and are thus prone to topological errors, e.g., missing or incorrectly merged/separated glands or nuclei. To address this issue, we propose TopoSemiSeg, the first semi-supervised method that learns the topological representation from unlabeled data. In particular, we propose a topology-aware teacher-student approach in which the teacher and student networks learn shared topological representations. To achieve this, we introduce topological consistency loss, which contains signal consistency and noise removal losses to ensure the learned representation is robust and focuses on true topological signals. Extensive experiments on public pathology image datasets show the superiority of our method, especially on topology-wise evaluation metrics. Code is available at https://github.com/Melon-Xu/TopoSemiSeg.
Calibrating Uncertainty for Semi-Supervised Crowd Counting
Aug 19, 2023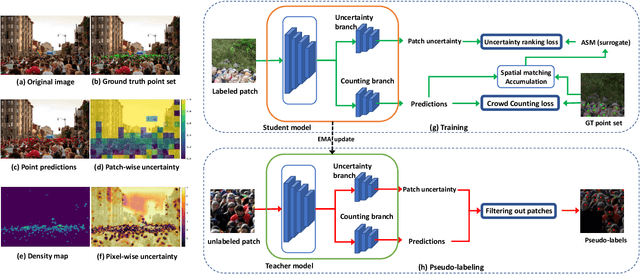

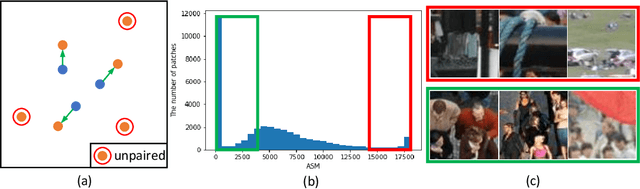

Semi-supervised crowd counting is an important yet challenging task. A popular approach is to iteratively generate pseudo-labels for unlabeled data and add them to the training set. The key is to use uncertainty to select reliable pseudo-labels. In this paper, we propose a novel method to calibrate model uncertainty for crowd counting. Our method takes a supervised uncertainty estimation strategy to train the model through a surrogate function. This ensures the uncertainty is well controlled throughout the training. We propose a matching-based patch-wise surrogate function to better approximate uncertainty for crowd counting tasks. The proposed method pays a sufficient amount of attention to details, while maintaining a proper granularity. Altogether our method is able to generate reliable uncertainty estimation, high quality pseudolabels, and achieve state-of-the-art performance in semisupervised crowd counting.
Topology-Guided Multi-Class Cell Context Generation for Digital Pathology
Apr 05, 2023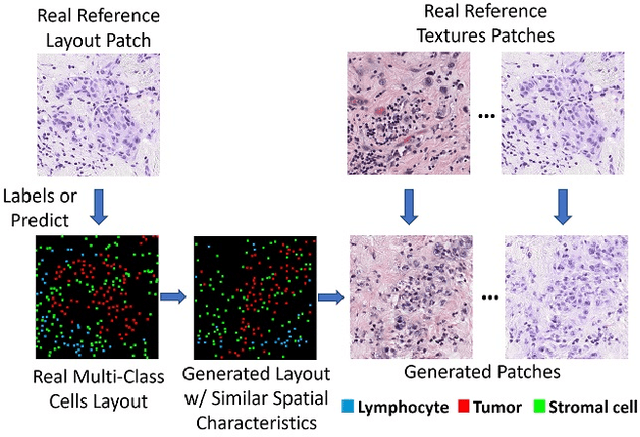

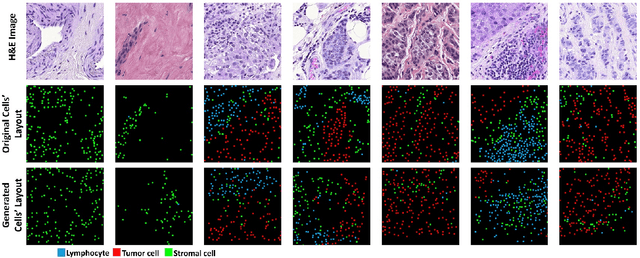

In digital pathology, the spatial context of cells is important for cell classification, cancer diagnosis and prognosis. To model such complex cell context, however, is challenging. Cells form different mixtures, lineages, clusters and holes. To model such structural patterns in a learnable fashion, we introduce several mathematical tools from spatial statistics and topological data analysis. We incorporate such structural descriptors into a deep generative model as both conditional inputs and a differentiable loss. This way, we are able to generate high quality multi-class cell layouts for the first time. We show that the topology-rich cell layouts can be used for data augmentation and improve the performance of downstream tasks such as cell classification.
Evaluating histopathology transfer learning with ChampKit
Jun 14, 2022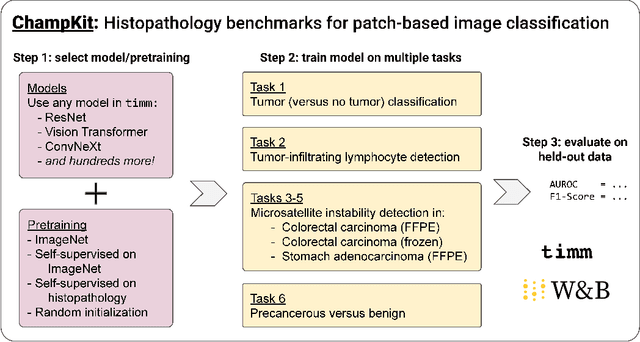
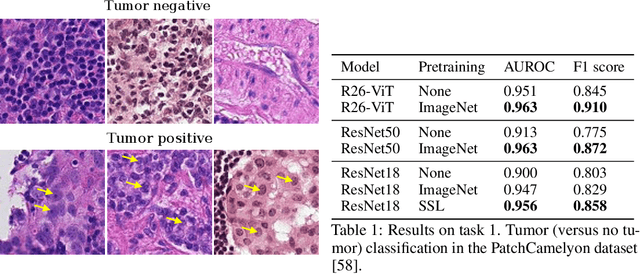
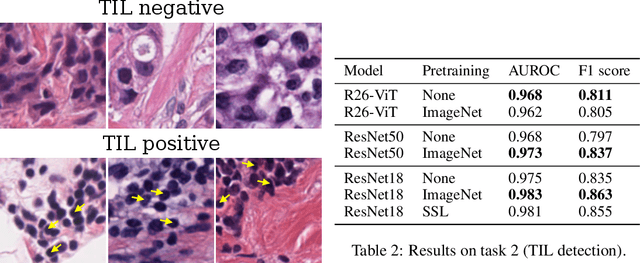
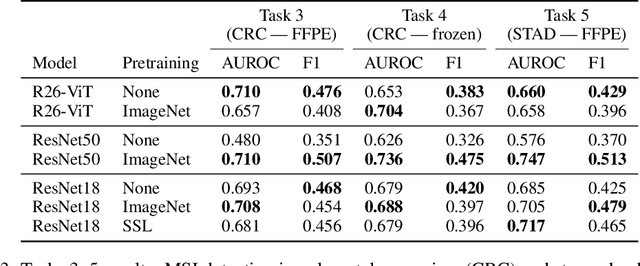
Histopathology remains the gold standard for diagnosis of various cancers. Recent advances in computer vision, specifically deep learning, have facilitated the analysis of histopathology images for various tasks, including immune cell detection and microsatellite instability classification. The state-of-the-art for each task often employs base architectures that have been pretrained for image classification on ImageNet. The standard approach to develop classifiers in histopathology tends to focus narrowly on optimizing models for a single task, not considering the aspects of modeling innovations that improve generalization across tasks. Here we present ChampKit (Comprehensive Histopathology Assessment of Model Predictions toolKit): an extensible, fully reproducible benchmarking toolkit that consists of a broad collection of patch-level image classification tasks across different cancers. ChampKit enables a way to systematically document the performance impact of proposed improvements in models and methodology. ChampKit source code and data are freely accessible at https://github.com/kaczmarj/champkit .
A Novel Framework for Characterization of Tumor-Immune Spatial Relationships in Tumor Microenvironment
May 01, 2022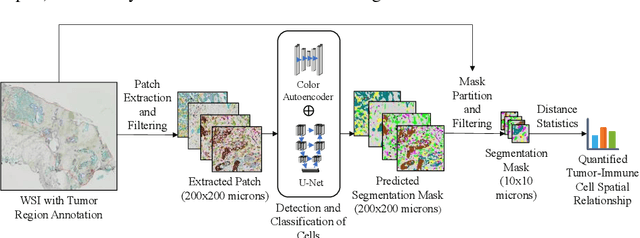

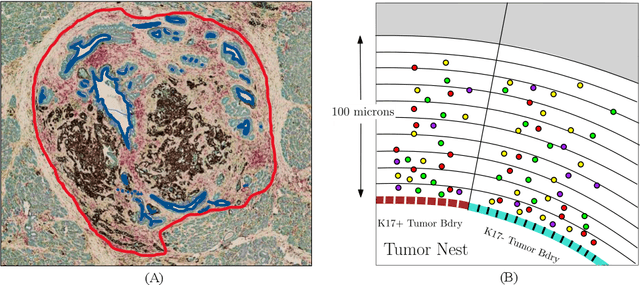

Understanding the impact of tumor biology on the composition of nearby cells often requires characterizing the impact of biologically distinct tumor regions. Biomarkers have been developed to label biologically distinct tumor regions, but challenges arise because of differences in the spatial extent and distribution of differentially labeled regions. In this work, we present a framework for systematically investigating the impact of distinct tumor regions on cells near the tumor borders, accounting their cross spatial distributions. We apply the framework to multiplex immunohistochemistry (mIHC) studies of pancreatic cancer and show its efficacy in demonstrating how biologically different tumor regions impact the immune response in the tumor microenvironment. Furthermore, we show that the proposed framework can be extended to largescale whole slide image analysis.
Federated Learning for the Classification of Tumor Infiltrating Lymphocytes
Apr 01, 2022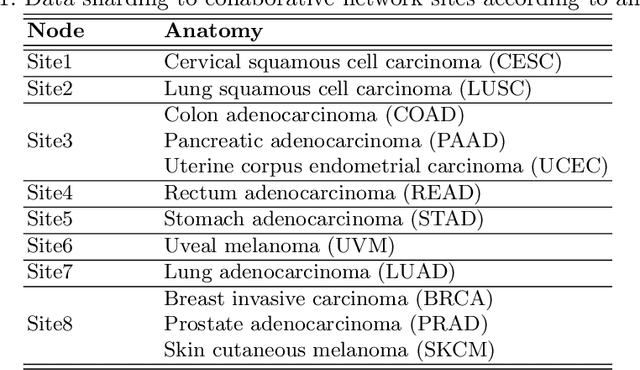

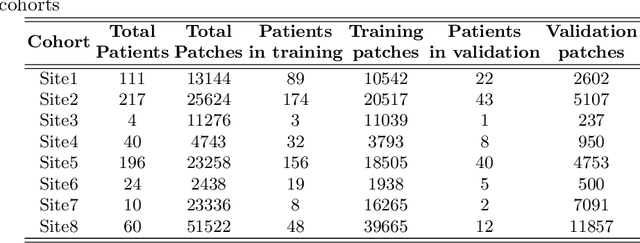

We evaluate the performance of federated learning (FL) in developing deep learning models for analysis of digitized tissue sections. A classification application was considered as the example use case, on quantifiying the distribution of tumor infiltrating lymphocytes within whole slide images (WSIs). A deep learning classification model was trained using 50*50 square micron patches extracted from the WSIs. We simulated a FL environment in which a dataset, generated from WSIs of cancer from numerous anatomical sites available by The Cancer Genome Atlas repository, is partitioned in 8 different nodes. Our results show that the model trained with the federated training approach achieves similar performance, both quantitatively and qualitatively, to that of a model trained with all the training data pooled at a centralized location. Our study shows that FL has tremendous potential for enabling development of more robust and accurate models for histopathology image analysis without having to collect large and diverse training data at a single location.
Multi-Class Cell Detection Using Spatial Context Representation
Oct 10, 2021

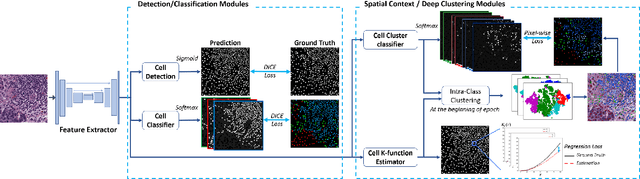
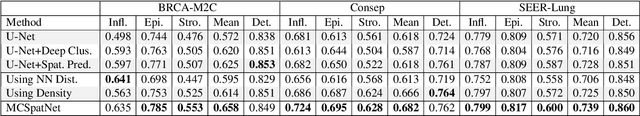
In digital pathology, both detection and classification of cells are important for automatic diagnostic and prognostic tasks. Classifying cells into subtypes, such as tumor cells, lymphocytes or stromal cells is particularly challenging. Existing methods focus on morphological appearance of individual cells, whereas in practice pathologists often infer cell classes through their spatial context. In this paper, we propose a novel method for both detection and classification that explicitly incorporates spatial contextual information. We use the spatial statistical function to describe local density in both a multi-class and a multi-scale manner. Through representation learning and deep clustering techniques, we learn advanced cell representation with both appearance and spatial context. On various benchmarks, our method achieves better performance than state-of-the-arts, especially on the classification task.
Localization in the Crowd with Topological Constraints
Dec 23, 2020


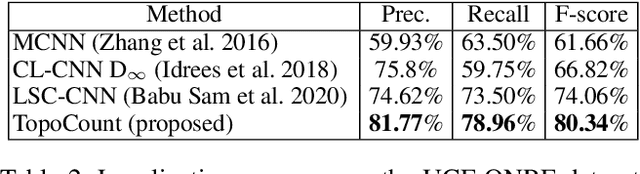
We address the problem of crowd localization, i.e., the prediction of dots corresponding to people in a crowded scene. Due to various challenges, a localization method is prone to spatial semantic errors, i.e., predicting multiple dots within a same person or collapsing multiple dots in a cluttered region. We propose a topological approach targeting these semantic errors. We introduce a topological constraint that teaches the model to reason about the spatial arrangement of dots. To enforce this constraint, we define a persistence loss based on the theory of persistent homology. The loss compares the topographic landscape of the likelihood map and the topology of the ground truth. Topological reasoning improves the quality of the localization algorithm especially near cluttered regions. On multiple public benchmarks, our method outperforms previous localization methods. Additionally, we demonstrate the potential of our method in improving the performance in the crowd counting task.
Exascale Deep Learning to Accelerate Cancer Research
Sep 26, 2019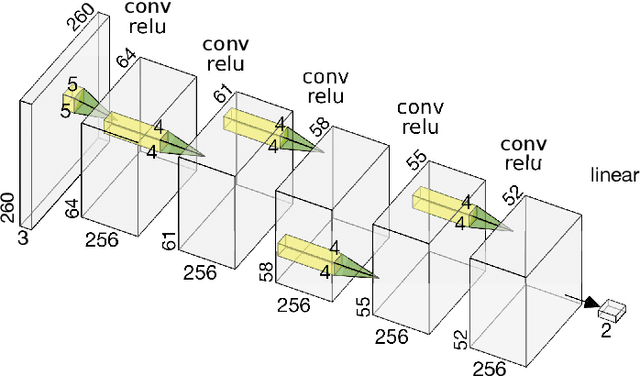
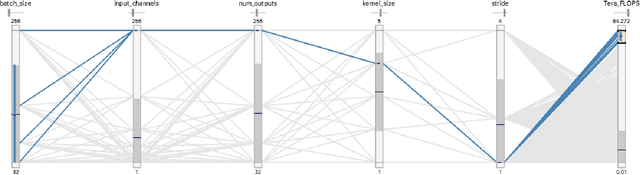
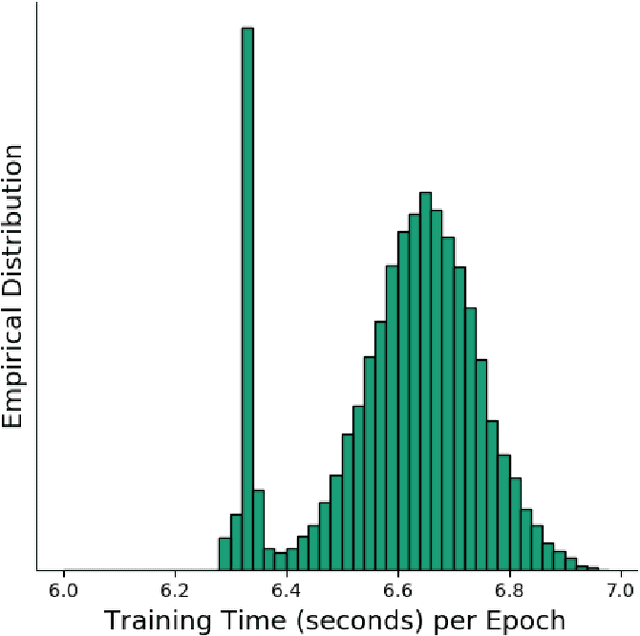
Deep learning, through the use of neural networks, has demonstrated remarkable ability to automate many routine tasks when presented with sufficient data for training. The neural network architecture (e.g. number of layers, types of layers, connections between layers, etc.) plays a critical role in determining what, if anything, the neural network is able to learn from the training data. The trend for neural network architectures, especially those trained on ImageNet, has been to grow ever deeper and more complex. The result has been ever increasing accuracy on benchmark datasets with the cost of increased computational demands. In this paper we demonstrate that neural network architectures can be automatically generated, tailored for a specific application, with dual objectives: accuracy of prediction and speed of prediction. Using MENNDL--an HPC-enabled software stack for neural architecture search--we generate a neural network with comparable accuracy to state-of-the-art networks on a cancer pathology dataset that is also $16\times$ faster at inference. The speedup in inference is necessary because of the volume and velocity of cancer pathology data; specifically, the previous state-of-the-art networks are too slow for individual researchers without access to HPC systems to keep pace with the rate of data generation. Our new model enables researchers with modest computational resources to analyze newly generated data faster than it is collected.
Learning from Thresholds: Fully Automated Classification of Tumor Infiltrating Lymphocytes for Multiple Cancer Types
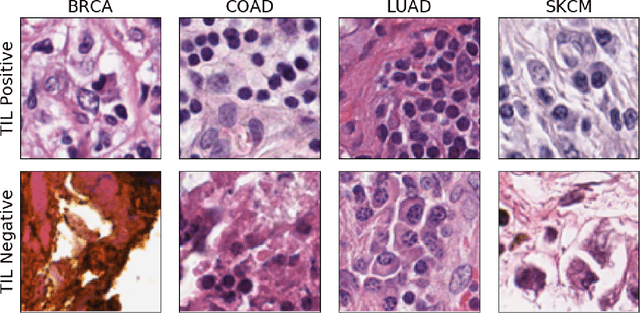
Jul 09, 2019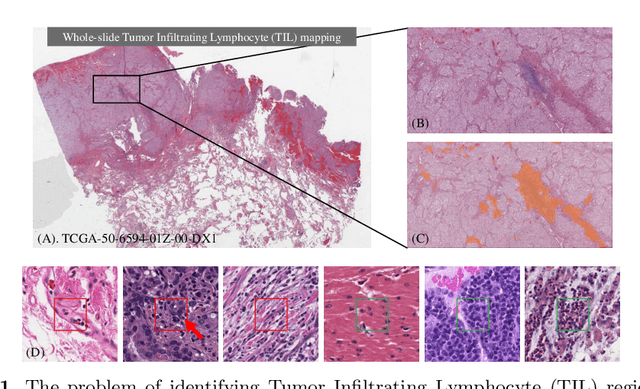
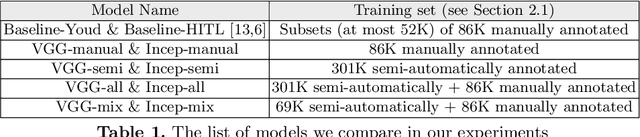

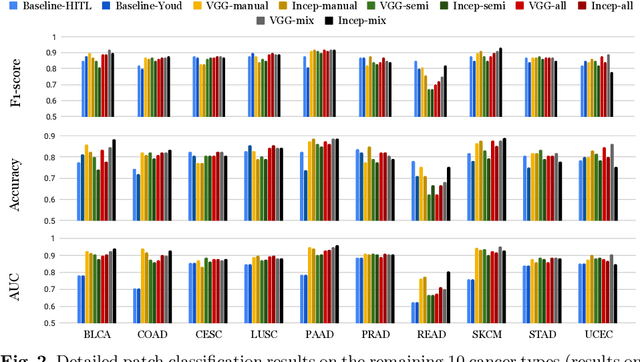
Deep learning classifiers for characterization of whole slide tissue morphology require large volumes of annotated data to learn variations across different tissue and cancer types. As is well known, manual generation of digital pathology training data is time consuming and expensive. In this paper, we propose a semi-automated method for annotating a group of similar instances at once, instead of collecting only per-instance manual annotations. This allows for a much larger training set, that reflects visual variability across multiple cancer types and thus training of a single network which can be automatically applied to each cancer type without human adjustment. We apply our method to the important task of classifying Tumor Infiltrating Lymphocytes (TILs) in H&E images. Prior approaches were trained for individual cancer types, with smaller training sets and human-in-the-loop threshold adjustment. We utilize these thresholded results as large scale "semi-automatic" annotations. Combined with existing manual annotations, our trained deep networks are able to automatically produce better TIL prediction results in 12 cancer types, compared to the human-in-the-loop approach.