 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeYikun Zhang
Atomas: Hierarchical Alignment on Molecule-Text for Unified Molecule Understanding and Generation
Apr 23, 2024
Molecule-and-text cross-modal representation learning has emerged as a promising direction for enhancing the quality of molecular representation, thereby improving performance in various scientific fields, including drug discovery and materials science. Existing studies adopt a global alignment approach to learn the knowledge from different modalities. These global alignment approaches fail to capture fine-grained information, such as molecular fragments and their corresponding textual description, which is crucial for downstream tasks. Furthermore, it is incapable to model such information using a similar global alignment strategy due to data scarcity of paired local part annotated data from existing datasets. In this paper, we propose Atomas, a multi-modal molecular representation learning framework to jointly learn representations from SMILES string and text. We design a Hierarchical Adaptive Alignment model to concurrently learn the fine-grained fragment correspondence between two modalities and align these representations of fragments in three levels. Additionally, Atomas's end-to-end training framework incorporates the tasks of understanding and generating molecule, thereby supporting a wider range of downstream tasks. In the retrieval task, Atomas exhibits robust generalization ability and outperforms the baseline by 30.8% of recall@1 on average. In the generation task, Atomas achieves state-of-the-art results in both molecule captioning task and molecule generation task. Moreover, the visualization of the Hierarchical Adaptive Alignment model further confirms the chemical significance of our approach. Our codes can be found at https://anonymous.4open.science/r/Atomas-03C3.
Functional Protein Design with Local Domain Alignment
Apr 18, 2024The core challenge of de novo protein design lies in creating proteins with specific functions or properties, guided by certain conditions. Current models explore to generate protein using structural and evolutionary guidance, which only provide indirect conditions concerning functions and properties. However, textual annotations of proteins, especially the annotations for protein domains, which directly describe the protein's high-level functionalities, properties, and their correlation with target amino acid sequences, remain unexplored in the context of protein design tasks. In this paper, we propose Protein-Annotation Alignment Generation (PAAG), a multi-modality protein design framework that integrates the textual annotations extracted from protein database for controllable generation in sequence space. Specifically, within a multi-level alignment module, PAAG can explicitly generate proteins containing specific domains conditioned on the corresponding domain annotations, and can even design novel proteins with flexible combinations of different kinds of annotations. Our experimental results underscore the superiority of the aligned protein representations from PAAG over 7 prediction tasks. Furthermore, PAAG demonstrates a nearly sixfold increase in generation success rate (24.7% vs 4.7% in zinc finger, and 54.3% vs 8.7% in the immunoglobulin domain) in comparison to the existing model.
Mode and Ridge Estimation in Euclidean and Directional Product Spaces: A Mean Shift Approach
Oct 16, 2021
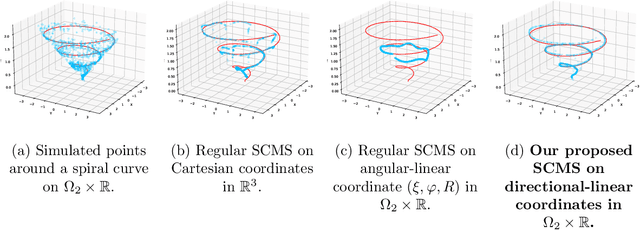
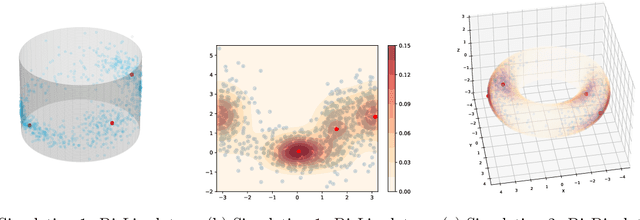
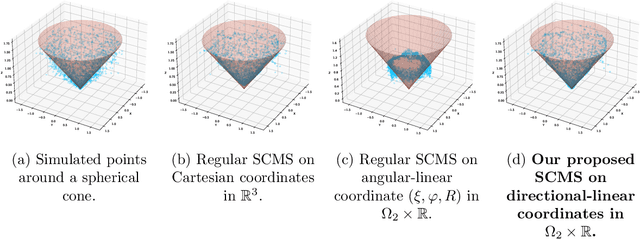
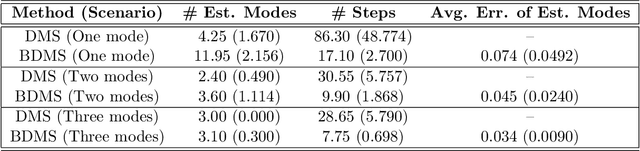
The set of local modes and the ridge lines estimated from a dataset are important summary characteristics of the data-generating distribution. In this work, we consider estimating the local modes and ridges from point cloud data in a product space with two or more Euclidean/directional metric spaces. Specifically, we generalize the well-known (subspace constrained) mean shift algorithm to the product space setting and illuminate some pitfalls in such generalization. We derive the algorithmic convergence of the proposed method, provide practical guidelines on the implementation, and demonstrate its effectiveness on both simulated and real datasets.
Linear Convergence of the Subspace Constrained Mean Shift Algorithm: From Euclidean to Directional Data
Apr 29, 2021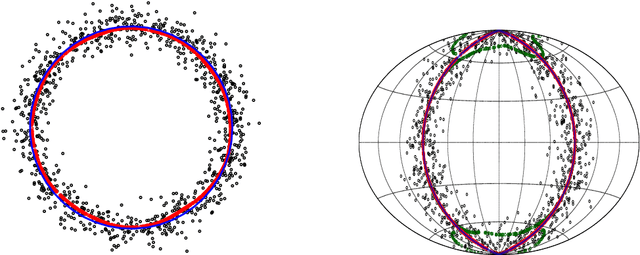

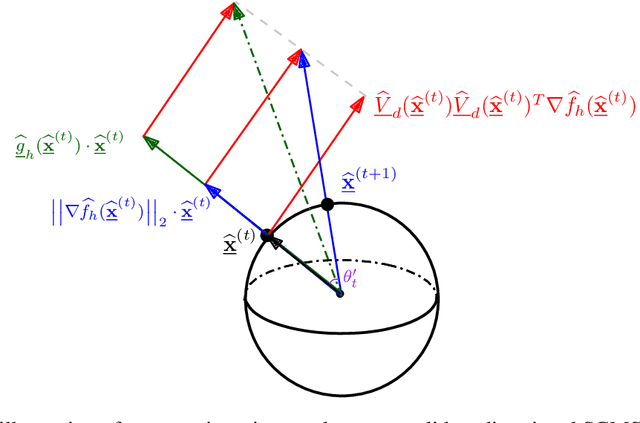
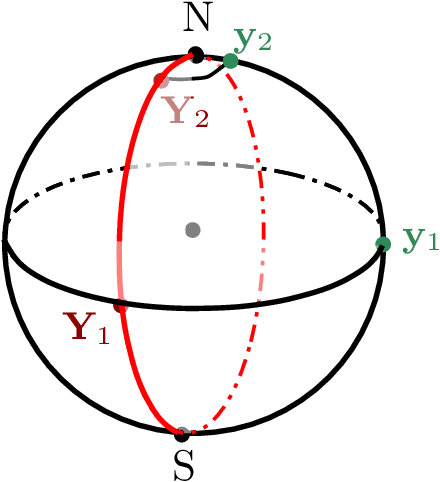
This paper studies linear convergence of the subspace constrained mean shift (SCMS) algorithm, a well-known algorithm for identifying a density ridge defined by a kernel density estimator. By arguing that the SCMS algorithm is a special variant of a subspace constrained gradient ascent (SCGA) algorithm with an adaptive step size, we derive linear convergence of such SCGA algorithm. While the existing research focuses mainly on density ridges in the Euclidean space, we generalize density ridges and the SCMS algorithm to directional data. In particular, we establish the stability theorem of density ridges with directional data and prove the linear convergence of our proposed directional SCMS algorithm.
The EM Perspective of Directional Mean Shift Algorithm
Jan 25, 2021
The directional mean shift (DMS) algorithm is a nonparametric method for pursuing local modes of densities defined by kernel density estimators on the unit hypersphere. In this paper, we show that any DMS iteration can be viewed as a generalized Expectation-Maximization (EM) algorithm; in particular, when the von Mises kernel is applied, it becomes an exact EM algorithm. Under the (generalized) EM framework, we provide a new proof for the ascending property of density estimates and demonstrate the global convergence of directional mean shift sequences. Finally, we give a new insight into the linear convergence of the DMS algorithm.
Kernel Smoothing, Mean Shift, and Their Learning Theory with Directional Data
Oct 23, 2020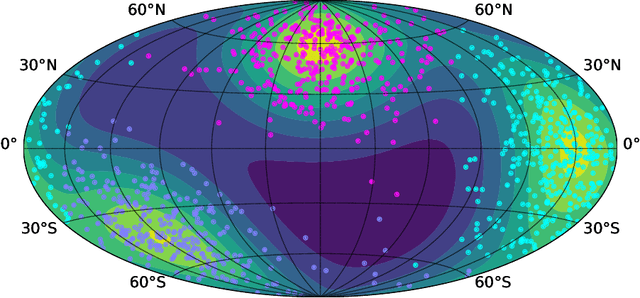
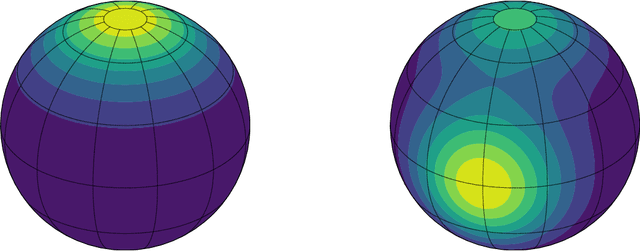
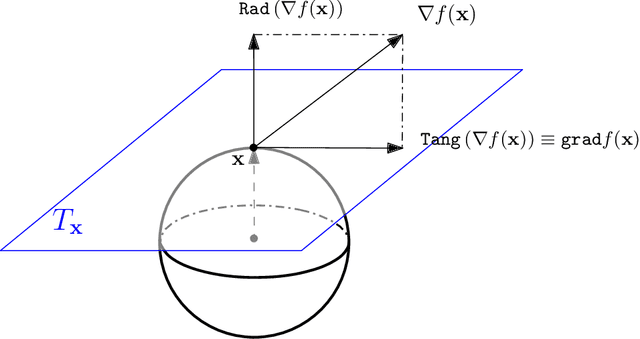
Directional data consist of observations distributed on a (hyper)sphere, and appear in many applied fields, such as astronomy, ecology, and environmental science. This paper studies both statistical and computational problems of kernel smoothing for directional data. We generalize the classical mean shift algorithm to directional data, which allows us to identify local modes of the directional kernel density estimator (KDE). The statistical convergence rates of the directional KDE and its derivatives are derived, and the problem of mode estimation is examined. We also prove the ascending property of our directional mean shift algorithm and investigate a general problem of gradient ascent on the unit hypersphere. To demonstrate the applicability of our proposed algorithm, we evaluate it as a mode clustering method on both simulated and real-world datasets.
Iterative Reconstruction for Low-Dose CT using Deep Gradient Priors of Generative Model
Sep 27, 2020



Dose reduction in computed tomography (CT) is essential for decreasing radiation risk in clinical applications. Iterative reconstruction is one of the most promising ways to compensate for the increased noise due to reduction of photon flux. Rather than most existing prior-driven algorithms that benefit from manually designed prior functions or supervised learning schemes, in this work we integrate the data-consistency as a conditional term into the iterative generative model for low-dose CT. At first, a score-based generative network is used for unsupervised distribution learning and the gradient of generative density prior is learned from normal-dose images. Then, the annealing Langevin dynamics is employed to update the trained priors with conditional scheme, i.e., the distance between the reconstructed image and the manifold is minimized along with data fidelity during reconstruction. Experimental comparisons demonstrated the noise reduction and detail preservation abilities of the proposed method.