 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeKaiyuan Gao
FABind+: Enhancing Molecular Docking through Improved Pocket Prediction and Pose Generation
Apr 07, 2024
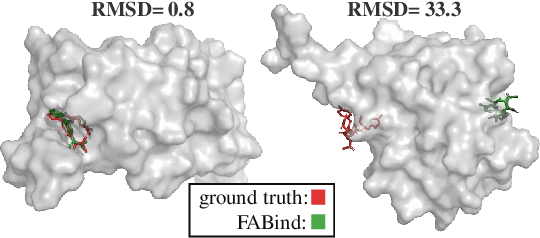
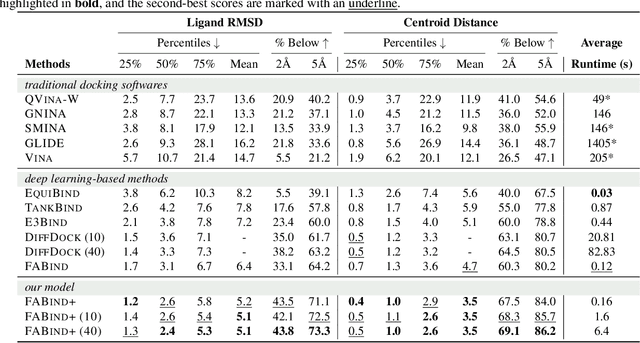
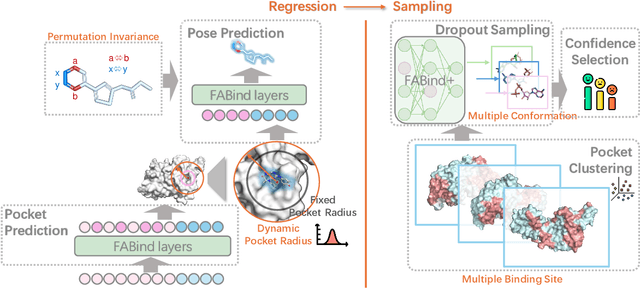
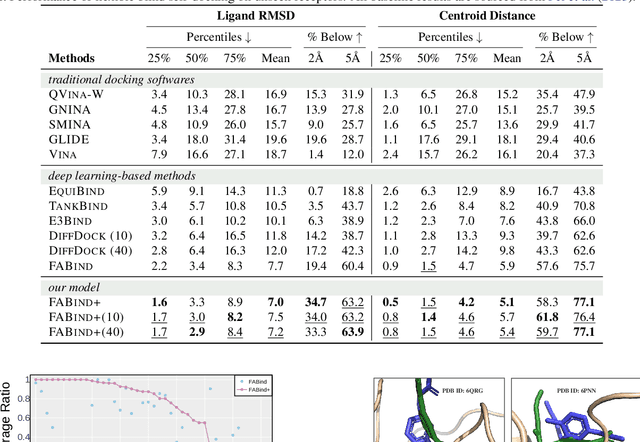
Molecular docking is a pivotal process in drug discovery. While traditional techniques rely on extensive sampling and simulation governed by physical principles, these methods are often slow and costly. The advent of deep learning-based approaches has shown significant promise, offering increases in both accuracy and efficiency. Building upon the foundational work of FABind, a model designed with a focus on speed and accuracy, we present FABind+, an enhanced iteration that largely boosts the performance of its predecessor. We identify pocket prediction as a critical bottleneck in molecular docking and propose a novel methodology that significantly refines pocket prediction, thereby streamlining the docking process. Furthermore, we introduce modifications to the docking module to enhance its pose generation capabilities. In an effort to bridge the gap with conventional sampling/generative methods, we incorporate a simple yet effective sampling technique coupled with a confidence model, requiring only minor adjustments to the regression framework of FABind. Experimental results and analysis reveal that FABind+ remarkably outperforms the original FABind, achieves competitive state-of-the-art performance, and delivers insightful modeling strategies. This demonstrates FABind+ represents a substantial step forward in molecular docking and drug discovery. Our code is in https://github.com/QizhiPei/FABind.
Leveraging Biomolecule and Natural Language through Multi-Modal Learning: A Survey
Mar 05, 2024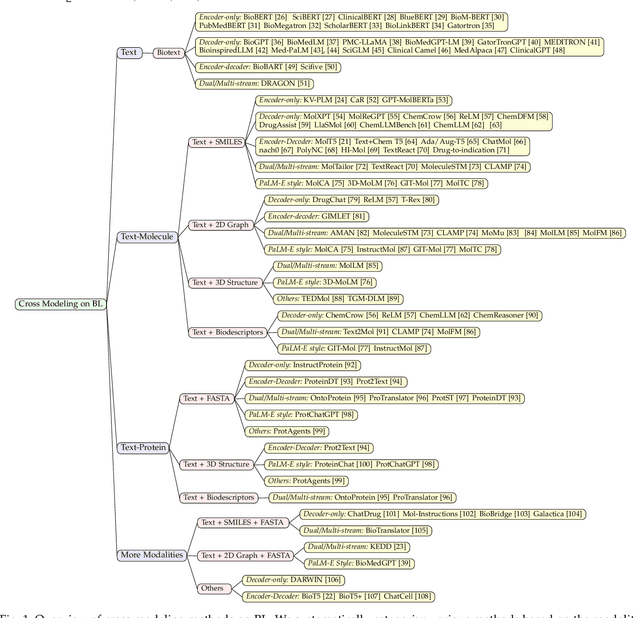
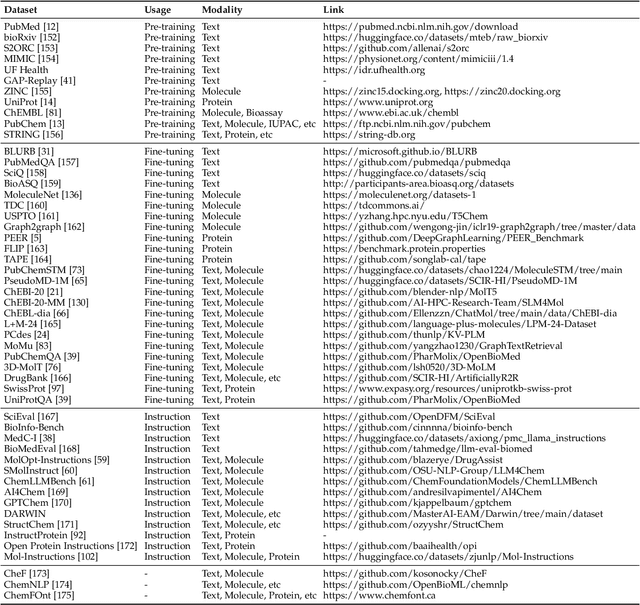
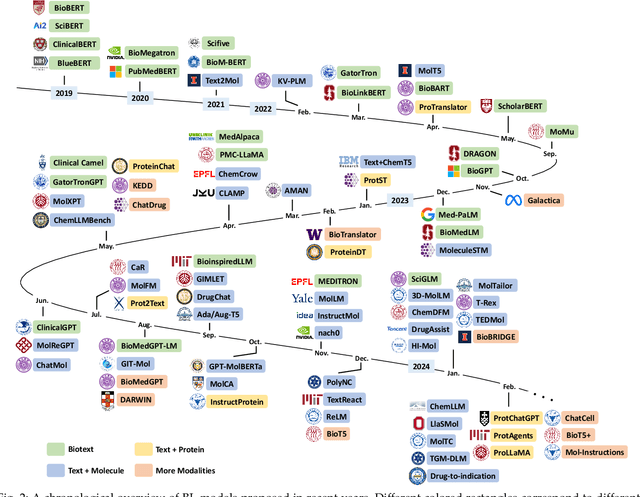
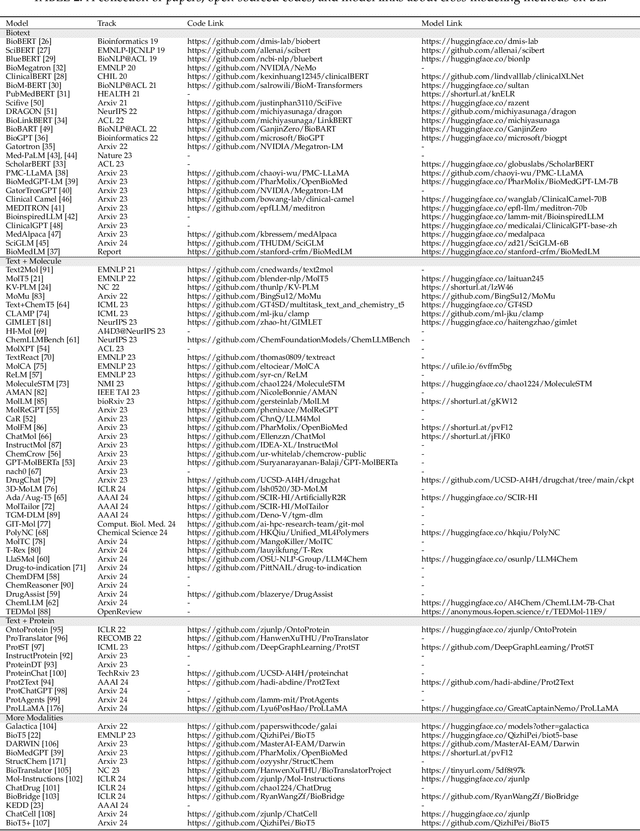
The integration of biomolecular modeling with natural language (BL) has emerged as a promising interdisciplinary area at the intersection of artificial intelligence, chemistry and biology. This approach leverages the rich, multifaceted descriptions of biomolecules contained within textual data sources to enhance our fundamental understanding and enable downstream computational tasks such as biomolecule property prediction. The fusion of the nuanced narratives expressed through natural language with the structural and functional specifics of biomolecules described via various molecular modeling techniques opens new avenues for comprehensively representing and analyzing biomolecules. By incorporating the contextual language data that surrounds biomolecules into their modeling, BL aims to capture a holistic view encompassing both the symbolic qualities conveyed through language as well as quantitative structural characteristics. In this review, we provide an extensive analysis of recent advancements achieved through cross modeling of biomolecules and natural language. (1) We begin by outlining the technical representations of biomolecules employed, including sequences, 2D graphs, and 3D structures. (2) We then examine in depth the rationale and key objectives underlying effective multi-modal integration of language and molecular data sources. (3) We subsequently survey the practical applications enabled to date in this developing research area. (4) We also compile and summarize the available resources and datasets to facilitate future work. (5) Looking ahead, we identify several promising research directions worthy of further exploration and investment to continue advancing the field. The related resources and contents are updating in \url{https://github.com/QizhiPei/Awesome-Biomolecule-Language-Cross-Modeling}.
BioT5+: Towards Generalized Biological Understanding with IUPAC Integration and Multi-task Tuning
Feb 27, 2024Recent research trends in computational biology have increasingly focused on integrating text and bio-entity modeling, especially in the context of molecules and proteins. However, previous efforts like BioT5 faced challenges in generalizing across diverse tasks and lacked a nuanced understanding of molecular structures, particularly in their textual representations (e.g., IUPAC). This paper introduces BioT5+, an extension of the BioT5 framework, tailored to enhance biological research and drug discovery. BioT5+ incorporates several novel features: integration of IUPAC names for molecular understanding, inclusion of extensive bio-text and molecule data from sources like bioRxiv and PubChem, the multi-task instruction tuning for generality across tasks, and a novel numerical tokenization technique for improved processing of numerical data. These enhancements allow BioT5+ to bridge the gap between molecular representations and their textual descriptions, providing a more holistic understanding of biological entities, and largely improving the grounded reasoning of bio-text and bio-sequences. The model is pre-trained and fine-tuned with a large number of experiments, including \emph{3 types of problems (classification, regression, generation), 15 kinds of tasks, and 21 total benchmark datasets}, demonstrating the remarkable performance and state-of-the-art results in most cases. BioT5+ stands out for its ability to capture intricate relationships in biological data, thereby contributing significantly to bioinformatics and computational biology. Our code is available at \url{https://github.com/QizhiPei/BioT5}.
BioT5: Enriching Cross-modal Integration in Biology with Chemical Knowledge and Natural Language Associations
Oct 17, 2023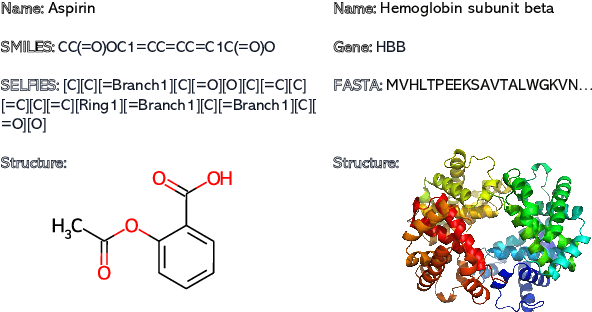
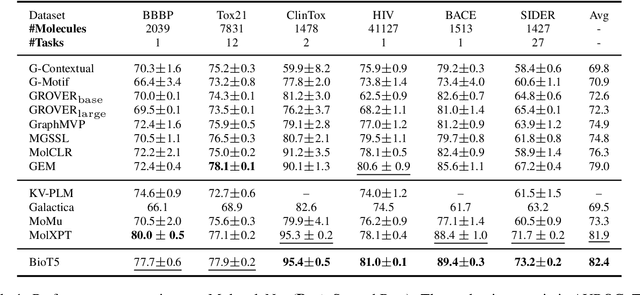
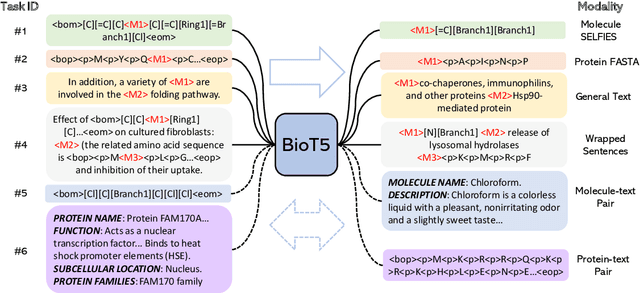
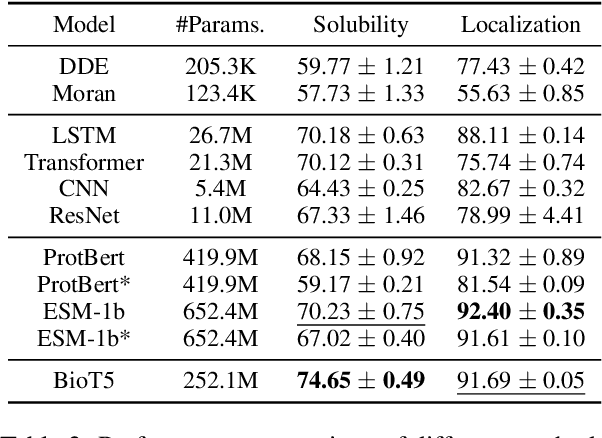
Recent advancements in biological research leverage the integration of molecules, proteins, and natural language to enhance drug discovery. However, current models exhibit several limitations, such as the generation of invalid molecular SMILES, underutilization of contextual information, and equal treatment of structured and unstructured knowledge. To address these issues, we propose $\mathbf{BioT5}$, a comprehensive pre-training framework that enriches cross-modal integration in biology with chemical knowledge and natural language associations. $\mathbf{BioT5}$ utilizes SELFIES for $100%$ robust molecular representations and extracts knowledge from the surrounding context of bio-entities in unstructured biological literature. Furthermore, $\mathbf{BioT5}$ distinguishes between structured and unstructured knowledge, leading to more effective utilization of information. After fine-tuning, BioT5 shows superior performance across a wide range of tasks, demonstrating its strong capability of capturing underlying relations and properties of bio-entities. Our code is available at $\href{https://github.com/QizhiPei/BioT5}{Github}$.
FABind: Fast and Accurate Protein-Ligand Binding
Oct 17, 2023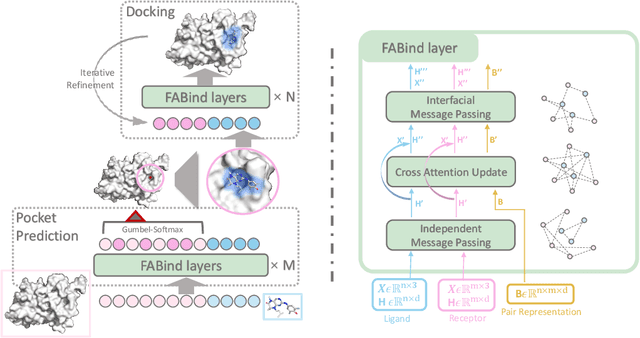
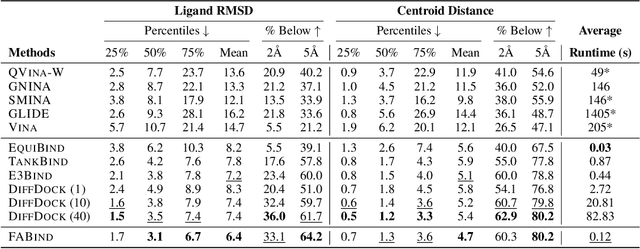
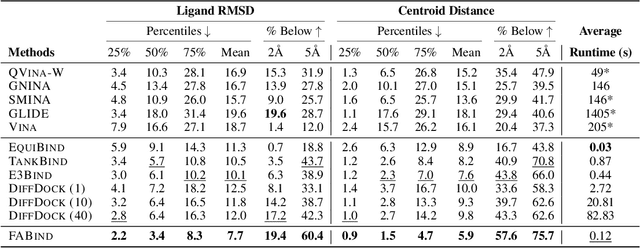
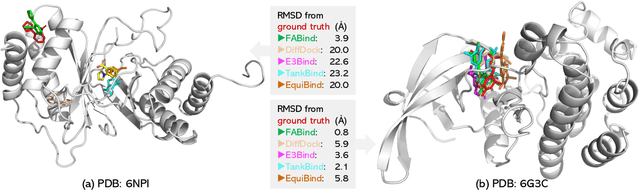
Modeling the interaction between proteins and ligands and accurately predicting their binding structures is a critical yet challenging task in drug discovery. Recent advancements in deep learning have shown promise in addressing this challenge, with sampling-based and regression-based methods emerging as two prominent approaches. However, these methods have notable limitations. Sampling-based methods often suffer from low efficiency due to the need for generating multiple candidate structures for selection. On the other hand, regression-based methods offer fast predictions but may experience decreased accuracy. Additionally, the variation in protein sizes often requires external modules for selecting suitable binding pockets, further impacting efficiency. In this work, we propose $\mathbf{FABind}$, an end-to-end model that combines pocket prediction and docking to achieve accurate and fast protein-ligand binding. $\mathbf{FABind}$ incorporates a unique ligand-informed pocket prediction module, which is also leveraged for docking pose estimation. The model further enhances the docking process by incrementally integrating the predicted pocket to optimize protein-ligand binding, reducing discrepancies between training and inference. Through extensive experiments on benchmark datasets, our proposed $\mathbf{FABind}$ demonstrates strong advantages in terms of effectiveness and efficiency compared to existing methods. Our code is available at $\href{https://github.com/QizhiPei/FABind}{Github}$.
Examining User-Friendly and Open-Sourced Large GPT Models: A Survey on Language, Multimodal, and Scientific GPT Models
Aug 27, 2023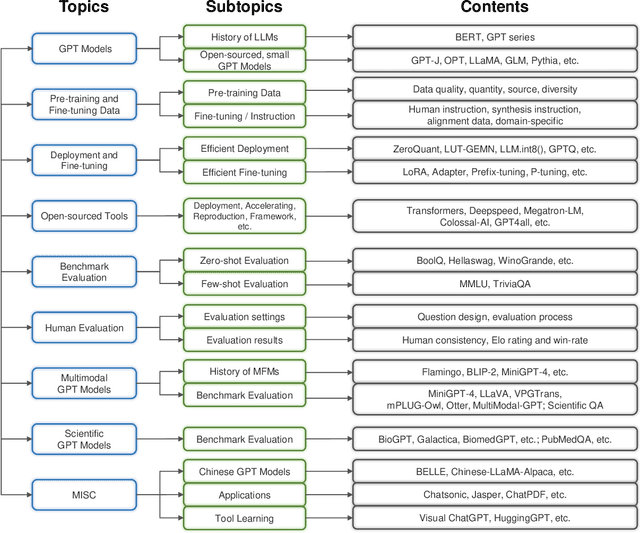
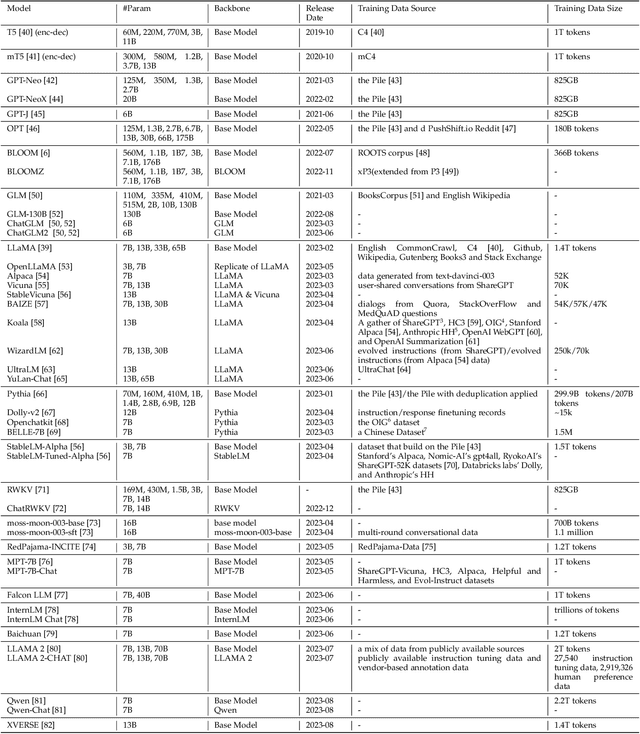
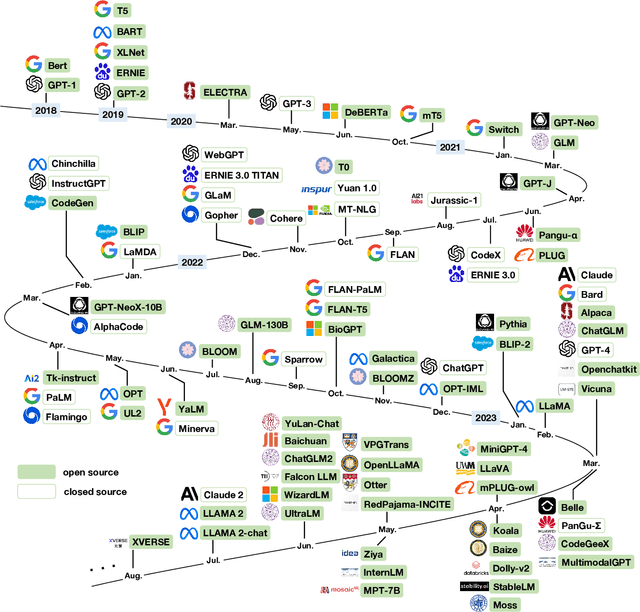
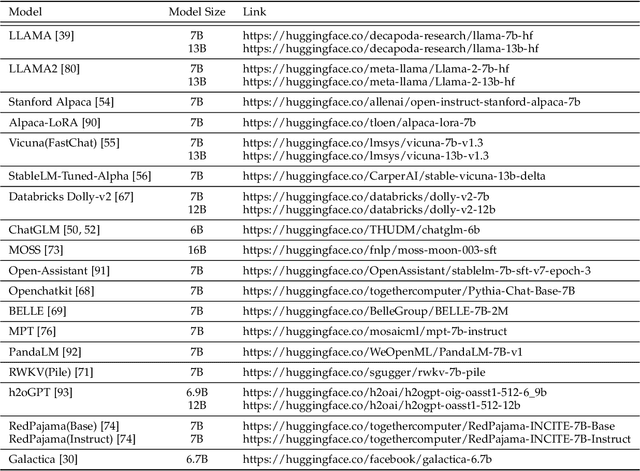
Generative pre-trained transformer (GPT) models have revolutionized the field of natural language processing (NLP) with remarkable performance in various tasks and also extend their power to multimodal domains. Despite their success, large GPT models like GPT-4 face inherent limitations such as considerable size, high computational requirements, complex deployment processes, and closed development loops. These constraints restrict their widespread adoption and raise concerns regarding their responsible development and usage. The need for user-friendly, relatively small, and open-sourced alternative GPT models arises from the desire to overcome these limitations while retaining high performance. In this survey paper, we provide an examination of alternative open-sourced models of large GPTs, focusing on user-friendly and relatively small models that facilitate easier deployment and accessibility. Through this extensive survey, we aim to equip researchers, practitioners, and enthusiasts with a thorough understanding of user-friendly and relatively small open-sourced models of large GPTs, their current state, challenges, and future research directions, inspiring the development of more efficient, accessible, and versatile GPT models that cater to the broader scientific community and advance the field of general artificial intelligence. The source contents are continuously updating in https://github.com/GPT-Alternatives/gpt_alternatives.
Tokenized Graph Transformer with Neighborhood Augmentation for Node Classification in Large Graphs
May 22, 2023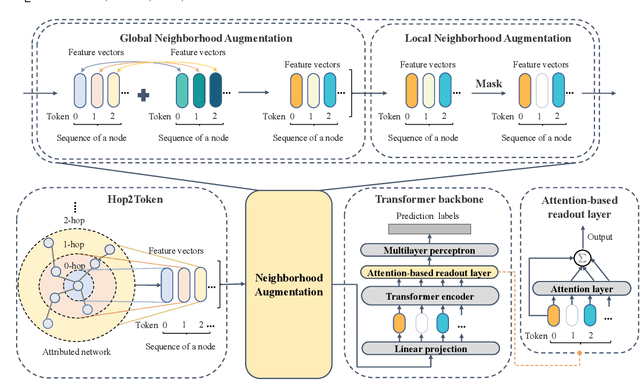
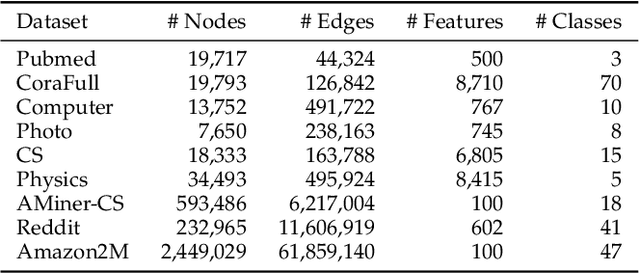
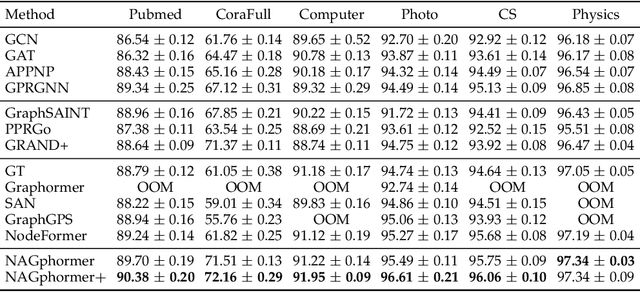

Graph Transformers, emerging as a new architecture for graph representation learning, suffer from the quadratic complexity on the number of nodes when handling large graphs. To this end, we propose a Neighborhood Aggregation Graph Transformer (NAGphormer) that treats each node as a sequence containing a series of tokens constructed by our proposed Hop2Token module. For each node, Hop2Token aggregates the neighborhood features from different hops into different representations, producing a sequence of token vectors as one input. In this way, NAGphormer could be trained in a mini-batch manner and thus could scale to large graphs. Moreover, we mathematically show that compared to a category of advanced Graph Neural Networks (GNNs), called decoupled Graph Convolutional Networks, NAGphormer could learn more informative node representations from multi-hop neighborhoods. In addition, we propose a new data augmentation method called Neighborhood Augmentation (NrAug) based on the output of Hop2Token that augments simultaneously the features of neighborhoods from global as well as local views to strengthen the training effect of NAGphormer. Extensive experiments on benchmark datasets from small to large demonstrate the superiority of NAGphormer against existing graph Transformers and mainstream GNNs, and the effectiveness of NrAug for further boosting NAGphormer.
Incorporating Pre-training Paradigm for Antibody Sequence-Structure Co-design
Nov 17, 2022Antibodies are versatile proteins that can bind to pathogens and provide effective protection for human body. Recently, deep learning-based computational antibody design has attracted popular attention since it automatically mines the antibody patterns from data that could be complementary to human experiences. However, the computational methods heavily rely on high-quality antibody structure data, which is quite limited. Besides, the complementarity-determining region (CDR), which is the key component of an antibody that determines the specificity and binding affinity, is highly variable and hard to predict. Therefore, the data limitation issue further raises the difficulty of CDR generation for antibodies. Fortunately, there exists a large amount of sequence data of antibodies that can help model the CDR and alleviate the reliance on structure data. By witnessing the success of pre-training models for protein modeling, in this paper, we develop the antibody pre-training language model and incorporate it into the (antigen-specific) antibody design model in a systemic way. Specifically, we first pre-train an antibody language model based on the sequence data, then propose a one-shot way for sequence and structure generation of CDR to avoid the heavy cost and error propagation from an autoregressive manner, and finally leverage the pre-trained antibody model for the antigen-specific antibody generation model with some carefully designed modules. Through various experiments, we show that our method achieves superior performances over previous baselines on different tasks, such as sequence and structure generation and antigen-binding CDR-H3 design.
NAGphormer: Neighborhood Aggregation Graph Transformer for Node Classification in Large Graphs
Jun 10, 2022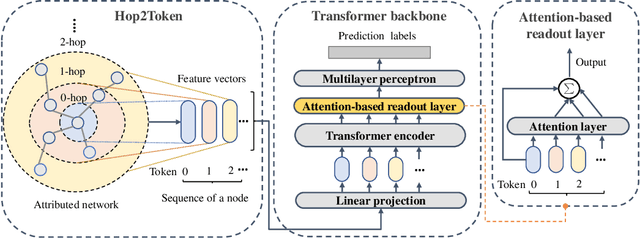
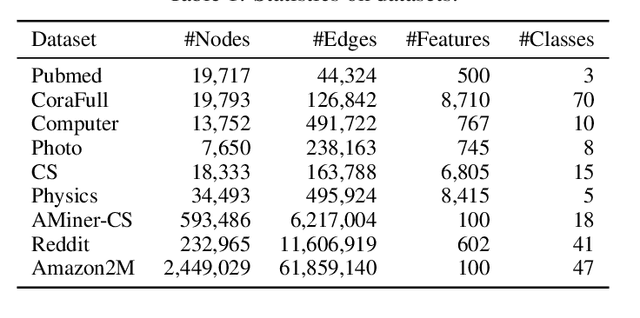
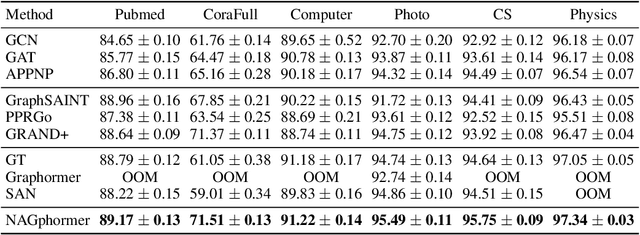
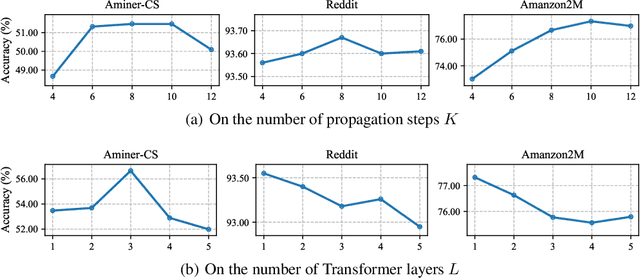
Graph Transformers have demonstrated superiority on various graph learning tasks in recent years. However, the complexity of existing Graph Transformers scales quadratically with the number of nodes, making it hard to scale to graphs with thousands of nodes. To this end, we propose a Neighborhood Aggregation Graph Transformer (NAGphormer) that is scalable to large graphs with millions of nodes. Before feeding the node features into the Transformer model, NAGphormer constructs tokens for each node by a neighborhood aggregation module called Hop2Token. For each node, Hop2Token aggregates neighborhood features from each hop into a representation, and thereby produces a sequence of token vectors. Subsequently, the resulting sequence of different hop information serves as input to the Transformer model. By considering each node as a sequence, NAGphormer could be trained in a mini-batch manner and thus could scale to large graphs. NAGphormer further develops an attention-based readout function so as to learn the importance of each hop adaptively. We conduct extensive experiments on various popular benchmarks, including six small datasets and three large datasets. The results demonstrate that NAGphormer consistently outperforms existing Graph Transformers and mainstream Graph Neural Networks.
Revisiting Language Encoding in Learning Multilingual Representations
Feb 16, 2021



Transformer has demonstrated its great power to learn contextual word representations for multiple languages in a single model. To process multilingual sentences in the model, a learnable vector is usually assigned to each language, which is called "language embedding". The language embedding can be either added to the word embedding or attached at the beginning of the sentence. It serves as a language-specific signal for the Transformer to capture contextual representations across languages. In this paper, we revisit the use of language embedding and identify several problems in the existing formulations. By investigating the interaction between language embedding and word embedding in the self-attention module, we find that the current methods cannot reflect the language-specific word correlation well. Given these findings, we propose a new approach called Cross-lingual Language Projection (XLP) to replace language embedding. For a sentence, XLP projects the word embeddings into language-specific semantic space, and then the projected embeddings will be fed into the Transformer model to process with their language-specific meanings. In such a way, XLP achieves the purpose of appropriately encoding "language" in a multilingual Transformer model. Experimental results show that XLP can freely and significantly boost the model performance on extensive multilingual benchmark datasets. Codes and models will be released at https://github.com/lsj2408/XLP.