 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeYuyan Ni
Contextual Molecule Representation Learning from Chemical Reaction Knowledge
Feb 21, 2024
In recent years, self-supervised learning has emerged as a powerful tool to harness abundant unlabelled data for representation learning and has been broadly adopted in diverse areas. However, when applied to molecular representation learning (MRL), prevailing techniques such as masked sub-unit reconstruction often fall short, due to the high degree of freedom in the possible combinations of atoms within molecules, which brings insurmountable complexity to the masking-reconstruction paradigm. To tackle this challenge, we introduce REMO, a self-supervised learning framework that takes advantage of well-defined atom-combination rules in common chemistry. Specifically, REMO pre-trains graph/Transformer encoders on 1.7 million known chemical reactions in the literature. We propose two pre-training objectives: Masked Reaction Centre Reconstruction (MRCR) and Reaction Centre Identification (RCI). REMO offers a novel solution to MRL by exploiting the underlying shared patterns in chemical reactions as \textit{context} for pre-training, which effectively infers meaningful representations of common chemistry knowledge. Such contextual representations can then be utilized to support diverse downstream molecular tasks with minimum finetuning, such as affinity prediction and drug-drug interaction prediction. Extensive experimental results on MoleculeACE, ACNet, drug-drug interaction (DDI), and reaction type classification show that across all tested downstream tasks, REMO outperforms the standard baseline of single-molecule masked modeling used in current MRL. Remarkably, REMO is the pioneering deep learning model surpassing fingerprint-based methods in activity cliff benchmarks.
Elastic Information Bottleneck
Nov 07, 2023Information bottleneck is an information-theoretic principle of representation learning that aims to learn a maximally compressed representation that preserves as much information about labels as possible. Under this principle, two different methods have been proposed, i.e., information bottleneck (IB) and deterministic information bottleneck (DIB), and have gained significant progress in explaining the representation mechanisms of deep learning algorithms. However, these theoretical and empirical successes are only valid with the assumption that training and test data are drawn from the same distribution, which is clearly not satisfied in many real-world applications. In this paper, we study their generalization abilities within a transfer learning scenario, where the target error could be decomposed into three components, i.e., source empirical error, source generalization gap (SG), and representation discrepancy (RD). Comparing IB and DIB on these terms, we prove that DIB's SG bound is tighter than IB's while DIB's RD is larger than IB's. Therefore, it is difficult to tell which one is better. To balance the trade-off between SG and the RD, we propose an elastic information bottleneck (EIB) to interpolate between the IB and DIB regularizers, which guarantees a Pareto frontier within the IB framework. Additionally, simulations and real data experiments show that EIB has the ability to achieve better domain adaptation results than IB and DIB, which validates the correctness of our theories.
Sliced Denoising: A Physics-Informed Molecular Pre-Training Method
Nov 03, 2023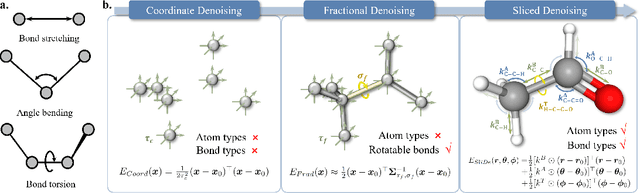

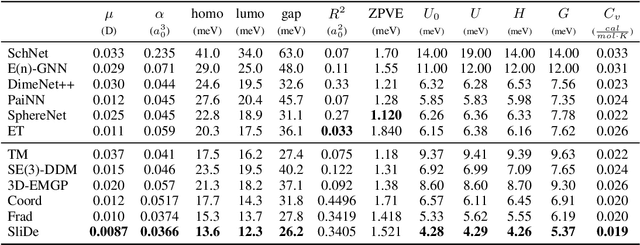
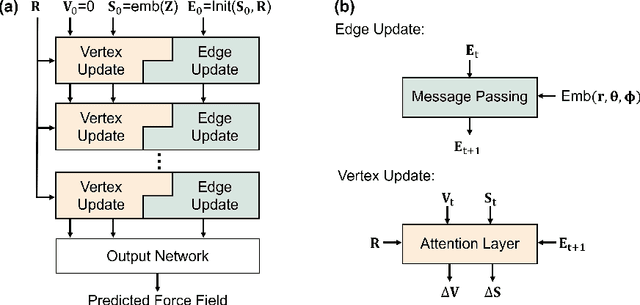
While molecular pre-training has shown great potential in enhancing drug discovery, the lack of a solid physical interpretation in current methods raises concerns about whether the learned representation truly captures the underlying explanatory factors in observed data, ultimately resulting in limited generalization and robustness. Although denoising methods offer a physical interpretation, their accuracy is often compromised by ad-hoc noise design, leading to inaccurate learned force fields. To address this limitation, this paper proposes a new method for molecular pre-training, called sliced denoising (SliDe), which is based on the classical mechanical intramolecular potential theory. SliDe utilizes a novel noise strategy that perturbs bond lengths, angles, and torsion angles to achieve better sampling over conformations. Additionally, it introduces a random slicing approach that circumvents the computationally expensive calculation of the Jacobian matrix, which is otherwise essential for estimating the force field. By aligning with physical principles, SliDe shows a 42\% improvement in the accuracy of estimated force fields compared to current state-of-the-art denoising methods, and thus outperforms traditional baselines on various molecular property prediction tasks.
Self-supervised Pocket Pretraining via Protein Fragment-Surroundings Alignment
Oct 11, 2023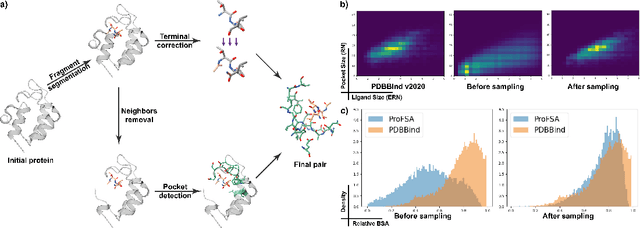
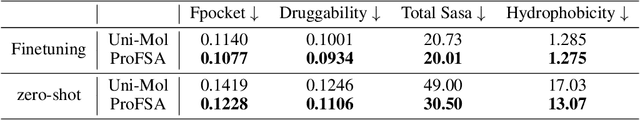
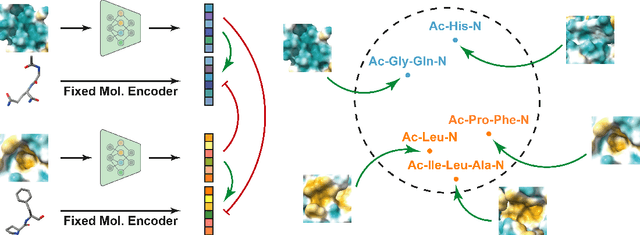
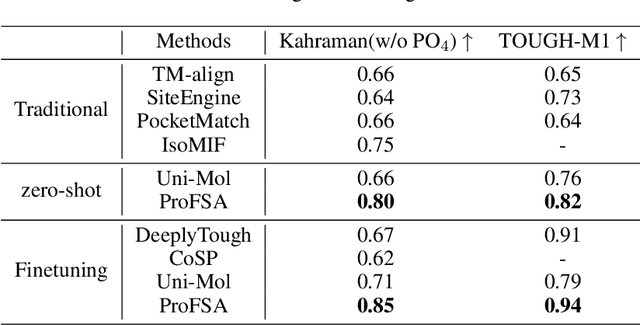
Pocket representations play a vital role in various biomedical applications, such as druggability estimation, ligand affinity prediction, and de novo drug design. While existing geometric features and pretrained representations have demonstrated promising results, they usually treat pockets independent of ligands, neglecting the fundamental interactions between them. However, the limited pocket-ligand complex structures available in the PDB database (less than 100 thousand non-redundant pairs) hampers large-scale pretraining endeavors for interaction modeling. To address this constraint, we propose a novel pocket pretraining approach that leverages knowledge from high-resolution atomic protein structures, assisted by highly effective pretrained small molecule representations. By segmenting protein structures into drug-like fragments and their corresponding pockets, we obtain a reasonable simulation of ligand-receptor interactions, resulting in the generation of over 5 million complexes. Subsequently, the pocket encoder is trained in a contrastive manner to align with the representation of pseudo-ligand furnished by some pretrained small molecule encoders. Our method, named ProFSA, achieves state-of-the-art performance across various tasks, including pocket druggability prediction, pocket matching, and ligand binding affinity prediction. Notably, ProFSA surpasses other pretraining methods by a substantial margin. Moreover, our work opens up a new avenue for mitigating the scarcity of protein-ligand complex data through the utilization of high-quality and diverse protein structure databases.
Fractional Denoising for 3D Molecular Pre-training
Jul 20, 2023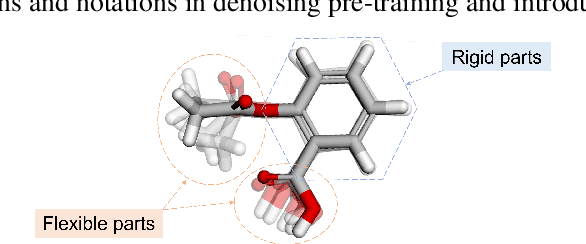

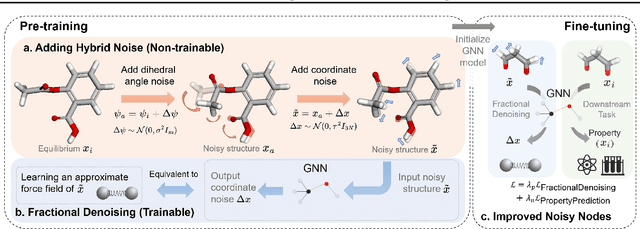
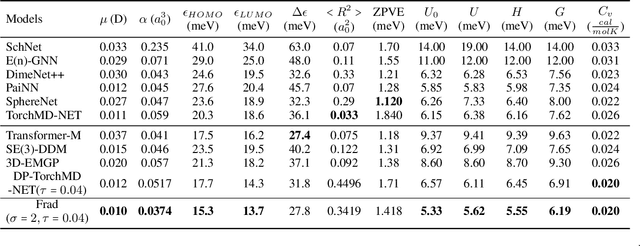
Coordinate denoising is a promising 3D molecular pre-training method, which has achieved remarkable performance in various downstream drug discovery tasks. Theoretically, the objective is equivalent to learning the force field, which is revealed helpful for downstream tasks. Nevertheless, there are two challenges for coordinate denoising to learn an effective force field, i.e. low coverage samples and isotropic force field. The underlying reason is that molecular distributions assumed by existing denoising methods fail to capture the anisotropic characteristic of molecules. To tackle these challenges, we propose a novel hybrid noise strategy, including noises on both dihedral angel and coordinate. However, denoising such hybrid noise in a traditional way is no more equivalent to learning the force field. Through theoretical deductions, we find that the problem is caused by the dependency of the input conformation for covariance. To this end, we propose to decouple the two types of noise and design a novel fractional denoising method (Frad), which only denoises the latter coordinate part. In this way, Frad enjoys both the merits of sampling more low-energy structures and the force field equivalence. Extensive experiments show the effectiveness of Frad in molecular representation, with a new state-of-the-art on 9 out of 12 tasks of QM9 and on 7 out of 8 targets of MD17.
Unified Molecular Modeling via Modality Blending
Jul 12, 2023
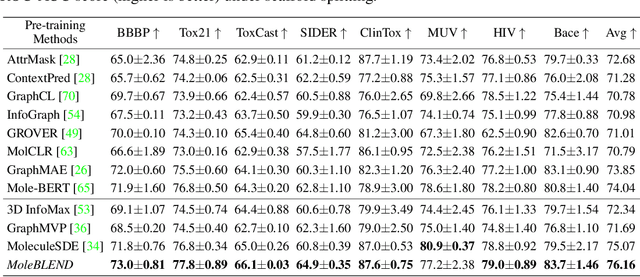

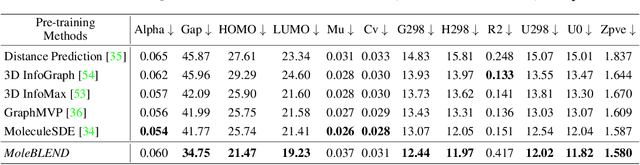
Self-supervised molecular representation learning is critical for molecule-based tasks such as AI-assisted drug discovery. Recent studies consider leveraging both 2D and 3D information for representation learning, with straightforward alignment strategies that treat each modality separately. In this work, we introduce a novel "blend-then-predict" self-supervised learning method (MoleBLEND), which blends atom relations from different modalities into one unified relation matrix for encoding, then recovers modality-specific information for both 2D and 3D structures. By treating atom relationships as anchors, seemingly dissimilar 2D and 3D manifolds are aligned and integrated at fine-grained relation-level organically. Extensive experiments show that MoleBLEND achieves state-of-the-art performance across major 2D/3D benchmarks. We further provide theoretical insights from the perspective of mutual-information maximization, demonstrating that our method unifies contrastive, generative (inter-modal prediction) and mask-then-predict (intra-modal prediction) objectives into a single cohesive blend-then-predict framework.