 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeZhi-Ming Ma
On the Convergence of Adam under Non-uniform Smoothness: Separability from SGDM and Beyond
Mar 22, 2024
This paper aims to clearly distinguish between Stochastic Gradient Descent with Momentum (SGDM) and Adam in terms of their convergence rates. We demonstrate that Adam achieves a faster convergence compared to SGDM under the condition of non-uniformly bounded smoothness. Our findings reveal that: (1) in deterministic environments, Adam can attain the known lower bound for the convergence rate of deterministic first-order optimizers, whereas the convergence rate of Gradient Descent with Momentum (GDM) has higher order dependence on the initial function value; (2) in stochastic setting, Adam's convergence rate upper bound matches the lower bounds of stochastic first-order optimizers, considering both the initial function value and the final error, whereas there are instances where SGDM fails to converge with any learning rate. These insights distinctly differentiate Adam and SGDM regarding their convergence rates. Additionally, by introducing a novel stopping-time based technique, we further prove that if we consider the minimum gradient norm during iterations, the corresponding convergence rate can match the lower bounds across all problem hyperparameters. The technique can also help proving that Adam with a specific hyperparameter scheduler is parameter-agnostic, which hence can be of independent interest.
The Surprising Effectiveness of Skip-Tuning in Diffusion Sampling
Feb 23, 2024With the incorporation of the UNet architecture, diffusion probabilistic models have become a dominant force in image generation tasks. One key design in UNet is the skip connections between the encoder and decoder blocks. Although skip connections have been shown to improve training stability and model performance, we reveal that such shortcuts can be a limiting factor for the complexity of the transformation. As the sampling steps decrease, the generation process and the role of the UNet get closer to the push-forward transformations from Gaussian distribution to the target, posing a challenge for the network's complexity. To address this challenge, we propose Skip-Tuning, a simple yet surprisingly effective training-free tuning method on the skip connections. Our method can achieve 100% FID improvement for pretrained EDM on ImageNet 64 with only 19 NFEs (1.75), breaking the limit of ODE samplers regardless of sampling steps. Surprisingly, the improvement persists when we increase the number of sampling steps and can even surpass the best result from EDM-2 (1.58) with only 39 NFEs (1.57). Comprehensive exploratory experiments are conducted to shed light on the surprising effectiveness. We observe that while Skip-Tuning increases the score-matching losses in the pixel space, the losses in the feature space are reduced, particularly at intermediate noise levels, which coincide with the most effective range accounting for image quality improvement.
Better Neural PDE Solvers Through Data-Free Mesh Movers
Dec 09, 2023Recently, neural networks have been extensively employed to solve partial differential equations (PDEs) in physical system modeling. While major studies focus on learning system evolution on predefined static mesh discretizations, some methods utilize reinforcement learning or supervised learning techniques to create adaptive and dynamic meshes, due to the dynamic nature of these systems. However, these approaches face two primary challenges: (1) the need for expensive optimal mesh data, and (2) the change of the solution space's degree of freedom and topology during mesh refinement. To address these challenges, this paper proposes a neural PDE solver with a neural mesh adapter. To begin with, we introduce a novel data-free neural mesh adaptor, called Data-free Mesh Mover (DMM), with two main innovations. Firstly, it is an operator that maps the solution to adaptive meshes and is trained using the Monge-Ampere equation without optimal mesh data. Secondly, it dynamically changes the mesh by moving existing nodes rather than adding or deleting nodes and edges. Theoretical analysis shows that meshes generated by DMM have the lowest interpolation error bound. Based on DMM, to efficiently and accurately model dynamic systems, we develop a moving mesh based neural PDE solver (MM-PDE) that embeds the moving mesh with a two-branch architecture and a learnable interpolation framework to preserve information within the data. Empirical experiments demonstrate that our method generates suitable meshes and considerably enhances accuracy when modeling widely considered PDE systems.
Deciphering and integrating invariants for neural operator learning with various physical mechanisms
Nov 24, 2023Neural operators have been explored as surrogate models for simulating physical systems to overcome the limitations of traditional partial differential equation (PDE) solvers. However, most existing operator learning methods assume that the data originate from a single physical mechanism, limiting their applicability and performance in more realistic scenarios. To this end, we propose Physical Invariant Attention Neural Operator (PIANO) to decipher and integrate the physical invariants (PI) for operator learning from the PDE series with various physical mechanisms. PIANO employs self-supervised learning to extract physical knowledge and attention mechanisms to integrate them into dynamic convolutional layers. Compared to existing techniques, PIANO can reduce the relative error by 13.6\%-82.2\% on PDE forecasting tasks across varying coefficients, forces, or boundary conditions. Additionally, varied downstream tasks reveal that the PI embeddings deciphered by PIANO align well with the underlying invariants in the PDE systems, verifying the physical significance of PIANO. The source code will be publicly available at: https://github.com/optray/PIANO.
Sliced Denoising: A Physics-Informed Molecular Pre-Training Method
Nov 03, 2023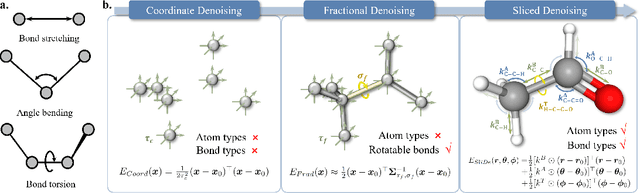

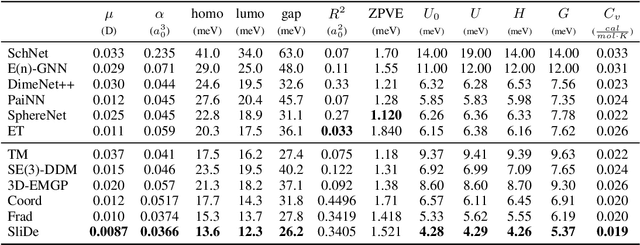
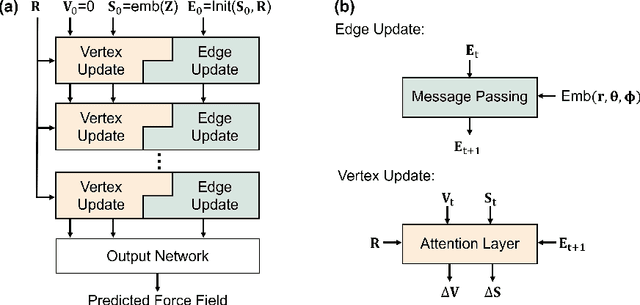
While molecular pre-training has shown great potential in enhancing drug discovery, the lack of a solid physical interpretation in current methods raises concerns about whether the learned representation truly captures the underlying explanatory factors in observed data, ultimately resulting in limited generalization and robustness. Although denoising methods offer a physical interpretation, their accuracy is often compromised by ad-hoc noise design, leading to inaccurate learned force fields. To address this limitation, this paper proposes a new method for molecular pre-training, called sliced denoising (SliDe), which is based on the classical mechanical intramolecular potential theory. SliDe utilizes a novel noise strategy that perturbs bond lengths, angles, and torsion angles to achieve better sampling over conformations. Additionally, it introduces a random slicing approach that circumvents the computationally expensive calculation of the Jacobian matrix, which is otherwise essential for estimating the force field. By aligning with physical principles, SliDe shows a 42\% improvement in the accuracy of estimated force fields compared to current state-of-the-art denoising methods, and thus outperforms traditional baselines on various molecular property prediction tasks.
SA-Solver: Stochastic Adams Solver for Fast Sampling of Diffusion Models
Sep 10, 2023
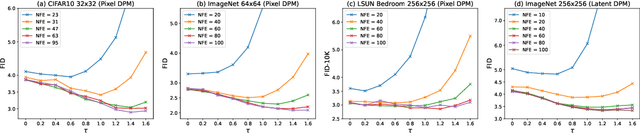

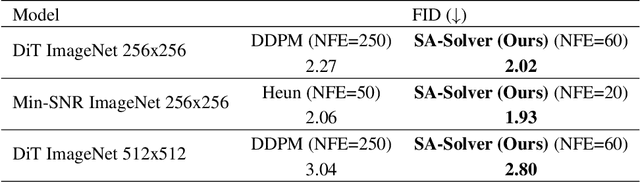
Diffusion Probabilistic Models (DPMs) have achieved considerable success in generation tasks. As sampling from DPMs is equivalent to solving diffusion SDE or ODE which is time-consuming, numerous fast sampling methods built upon improved differential equation solvers are proposed. The majority of such techniques consider solving the diffusion ODE due to its superior efficiency. However, stochastic sampling could offer additional advantages in generating diverse and high-quality data. In this work, we engage in a comprehensive analysis of stochastic sampling from two aspects: variance-controlled diffusion SDE and linear multi-step SDE solver. Based on our analysis, we propose SA-Solver, which is an improved efficient stochastic Adams method for solving diffusion SDE to generate data with high quality. Our experiments show that SA-Solver achieves: 1) improved or comparable performance compared with the existing state-of-the-art sampling methods for few-step sampling; 2) SOTA FID scores on substantial benchmark datasets under a suitable number of function evaluations (NFEs).
Fractional Denoising for 3D Molecular Pre-training
Jul 20, 2023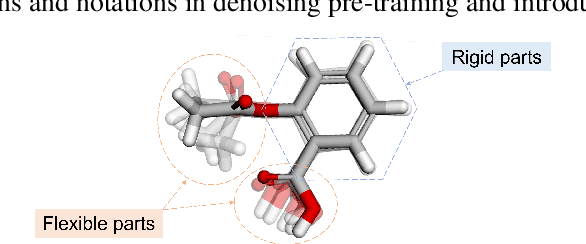

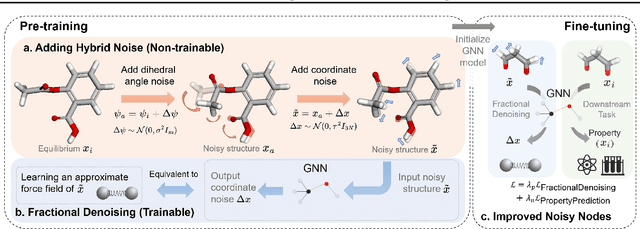
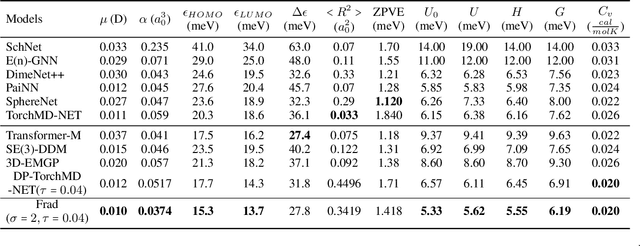
Coordinate denoising is a promising 3D molecular pre-training method, which has achieved remarkable performance in various downstream drug discovery tasks. Theoretically, the objective is equivalent to learning the force field, which is revealed helpful for downstream tasks. Nevertheless, there are two challenges for coordinate denoising to learn an effective force field, i.e. low coverage samples and isotropic force field. The underlying reason is that molecular distributions assumed by existing denoising methods fail to capture the anisotropic characteristic of molecules. To tackle these challenges, we propose a novel hybrid noise strategy, including noises on both dihedral angel and coordinate. However, denoising such hybrid noise in a traditional way is no more equivalent to learning the force field. Through theoretical deductions, we find that the problem is caused by the dependency of the input conformation for covariance. To this end, we propose to decouple the two types of noise and design a novel fractional denoising method (Frad), which only denoises the latter coordinate part. In this way, Frad enjoys both the merits of sampling more low-energy structures and the force field equivalence. Extensive experiments show the effectiveness of Frad in molecular representation, with a new state-of-the-art on 9 out of 12 tasks of QM9 and on 7 out of 8 targets of MD17.
Power-law Dynamic arising from machine learning
Jun 16, 2023We study a kind of new SDE that was arisen from the research on optimization in machine learning, we call it power-law dynamic because its stationary distribution cannot have sub-Gaussian tail and obeys power-law. We prove that the power-law dynamic is ergodic with unique stationary distribution, provided the learning rate is small enough. We investigate its first exist time. In particular, we compare the exit times of the (continuous) power-law dynamic and its discretization. The comparison can help guide machine learning algorithm.
Convergence of AdaGrad for Non-convex Objectives: Simple Proofs and Relaxed Assumptions
May 29, 2023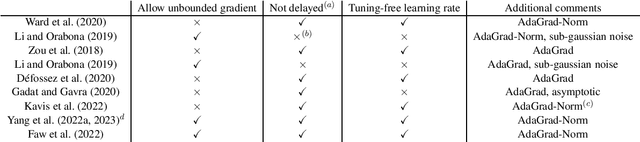
We provide a simple convergence proof for AdaGrad optimizing non-convex objectives under only affine noise variance and bounded smoothness assumptions. The proof is essentially based on a novel auxiliary function $\xi$ that helps eliminate the complexity of handling the correlation between the numerator and denominator of AdaGrad's update. Leveraging simple proofs, we are able to obtain tighter results than existing results \citep{faw2022power} and extend the analysis to several new and important cases. Specifically, for the over-parameterized regime, we show that AdaGrad needs only $\mathcal{O}(\frac{1}{\varepsilon^2})$ iterations to ensure the gradient norm smaller than $\varepsilon$, which matches the rate of SGD and significantly tighter than existing rates $\mathcal{O}(\frac{1}{\varepsilon^4})$ for AdaGrad. We then discard the bounded smoothness assumption and consider a realistic assumption on smoothness called $(L_0,L_1)$-smooth condition, which allows local smoothness to grow with the gradient norm. Again based on the auxiliary function $\xi$, we prove that AdaGrad succeeds in converging under $(L_0,L_1)$-smooth condition as long as the learning rate is lower than a threshold. Interestingly, we further show that the requirement on learning rate under the $(L_0,L_1)$-smooth condition is necessary via proof by contradiction, in contrast with the case of uniform smoothness conditions where convergence is guaranteed regardless of learning rate choices. Together, our analyses broaden the understanding of AdaGrad and demonstrate the power of the new auxiliary function in the investigations of AdaGrad.
A new perspective on building efficient and expressive 3D equivariant graph neural networks
Apr 07, 2023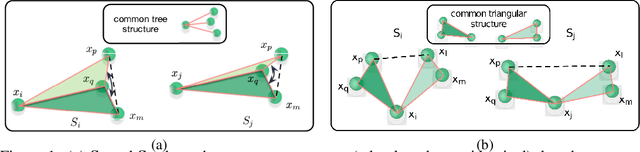
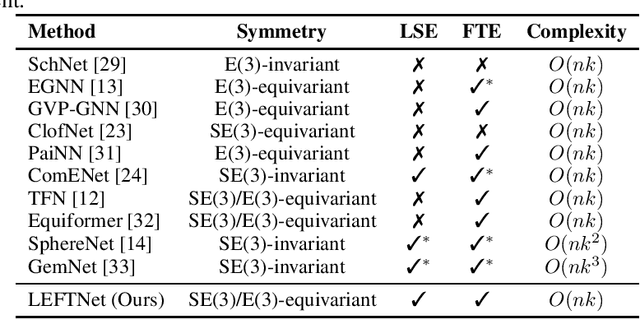

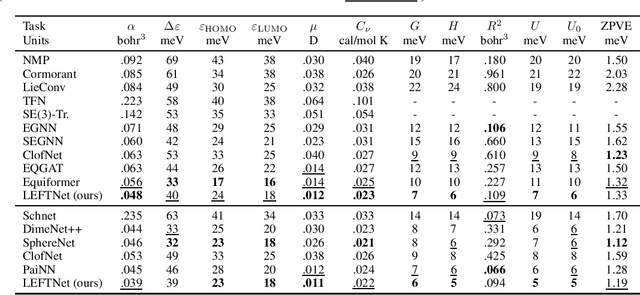
Geometric deep learning enables the encoding of physical symmetries in modeling 3D objects. Despite rapid progress in encoding 3D symmetries into Graph Neural Networks (GNNs), a comprehensive evaluation of the expressiveness of these networks through a local-to-global analysis lacks today. In this paper, we propose a local hierarchy of 3D isomorphism to evaluate the expressive power of equivariant GNNs and investigate the process of representing global geometric information from local patches. Our work leads to two crucial modules for designing expressive and efficient geometric GNNs; namely local substructure encoding (LSE) and frame transition encoding (FTE). To demonstrate the applicability of our theory, we propose LEFTNet which effectively implements these modules and achieves state-of-the-art performance on both scalar-valued and vector-valued molecular property prediction tasks. We further point out the design space for future developments of equivariant graph neural networks. Our codes are available at \url{https://github.com/yuanqidu/LeftNet}.