 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTianfan Fu
CT-Agent: Clinical Trial Multi-Agent with Large Language Model-based Reasoning
Apr 23, 2024
Large Language Models (LLMs) and multi-agent systems have shown impressive capabilities in natural language tasks but face challenges in clinical trial applications, primarily due to limited access to external knowledge. Recognizing the potential of advanced clinical trial tools that aggregate and predict based on the latest medical data, we propose an integrated solution to enhance their accessibility and utility. We introduce Clinical Agent System (CT-Agent), a Clinical multi-agent system designed for clinical trial tasks, leveraging GPT-4, multi-agent architectures, LEAST-TO-MOST, and ReAct reasoning technology. This integration not only boosts LLM performance in clinical contexts but also introduces novel functionalities. Our system autonomously manages the entire clinical trial process, demonstrating significant efficiency improvements in our evaluations, which include both computational benchmarks and expert feedback.
TrialDura: Hierarchical Attention Transformer for Interpretable Clinical Trial Duration Prediction
Apr 20, 2024The clinical trial process, also known as drug development, is an indispensable step toward the development of new treatments. The major objective of interventional clinical trials is to assess the safety and effectiveness of drug-based treatment in treating certain diseases in the human body. However, clinical trials are lengthy, labor-intensive, and costly. The duration of a clinical trial is a crucial factor that influences overall expenses. Therefore, effective management of the timeline of a clinical trial is essential for controlling the budget and maximizing the economic viability of the research. To address this issue, We propose TrialDura, a machine learning-based method that estimates the duration of clinical trials using multimodal data, including disease names, drug molecules, trial phases, and eligibility criteria. Then, we encode them into Bio-BERT embeddings specifically tuned for biomedical contexts to provide a deeper and more relevant semantic understanding of clinical trial data. Finally, the model's hierarchical attention mechanism connects all of the embeddings to capture their interactions and predict clinical trial duration. Our proposed model demonstrated superior performance with a mean absolute error (MAE) of 1.04 years and a root mean square error (RMSE) of 1.39 years compared to the other models, indicating more accurate clinical trial duration prediction. Publicly available code can be found at https://anonymous.4open.science/r/TrialDura-F196
AUTODIFF: Autoregressive Diffusion Modeling for Structure-based Drug Design
Apr 03, 2024Structure-based drug design (SBDD), which aims to generate molecules that can bind tightly to the target protein, is an essential problem in drug discovery, and previous approaches have achieved initial success. However, most existing methods still suffer from invalid local structure or unrealistic conformation issues, which are mainly due to the poor leaning of bond angles or torsional angles. To alleviate these problems, we propose AUTODIFF, a diffusion-based fragment-wise autoregressive generation model. Specifically, we design a novel molecule assembly strategy named conformal motif that preserves the conformation of local structures of molecules first, then we encode the interaction of the protein-ligand complex with an SE(3)-equivariant convolutional network and generate molecules motif-by-motif with diffusion modeling. In addition, we also improve the evaluation framework of SBDD by constraining the molecular weights of the generated molecules in the same range, together with some new metrics, which make the evaluation more fair and practical. Extensive experiments on CrossDocked2020 demonstrate that our approach outperforms the existing models in generating realistic molecules with valid structures and conformations while maintaining high binding affinity.
Multimodal Clinical Trial Outcome Prediction with Large Language Models
Feb 18, 2024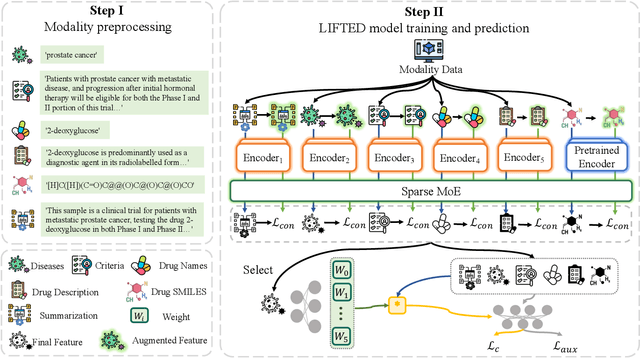
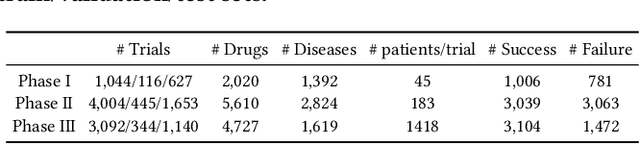
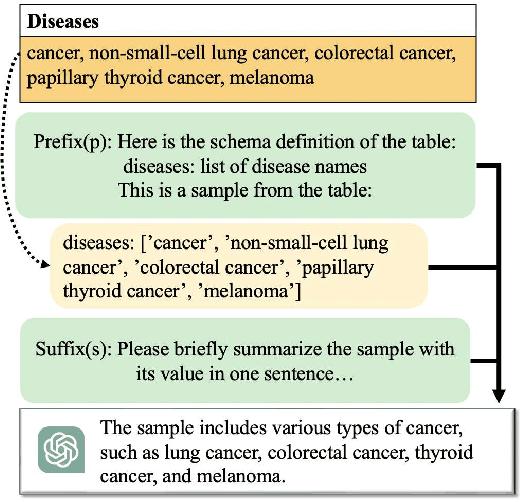
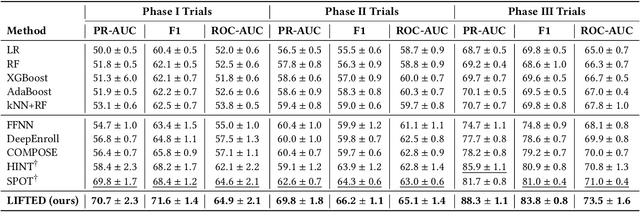
The clinical trial is a pivotal and costly process, often spanning multiple years and requiring substantial financial resources. Therefore, the development of clinical trial outcome prediction models aims to exclude drugs likely to fail and holds the potential for significant cost savings. Recent data-driven attempts leverage deep learning methods to integrate multimodal data for predicting clinical trial outcomes. However, these approaches rely on manually designed modal-specific encoders, which limits both the extensibility to adapt new modalities and the ability to discern similar information patterns across different modalities. To address these issues, we propose a multimodal mixture-of-experts (LIFTED) approach for clinical trial outcome prediction. Specifically, LIFTED unifies different modality data by transforming them into natural language descriptions. Then, LIFTED constructs unified noise-resilient encoders to extract information from modal-specific language descriptions. Subsequently, a sparse Mixture-of-Experts framework is employed to further refine the representations, enabling LIFTED to identify similar information patterns across different modalities and extract more consistent representations from those patterns using the same expert model. Finally, a mixture-of-experts module is further employed to dynamically integrate different modality representations for prediction, which gives LIFTED the ability to automatically weigh different modalities and pay more attention to critical information. The experiments demonstrate that LIFTED significantly enhances performance in predicting clinical trial outcomes across all three phases compared to the best baseline, showcasing the effectiveness of our proposed key components.
Molecular De Novo Design through Transformer-based Reinforcement Learning
Oct 10, 2023



In this work, we introduce a method to fine-tune a Transformer-based generative model for molecular de novo design. Leveraging the superior sequence learning capacity of Transformers over Recurrent Neural Networks (RNNs), our model can generate molecular structures with desired properties effectively. In contrast to the traditional RNN-based models, our proposed method exhibits superior performance in generating compounds predicted to be active against various biological targets, capturing long-term dependencies in the molecular structure sequence. The model's efficacy is demonstrated across numerous tasks, including generating analogues to a query structure and producing compounds with particular attributes, outperforming the baseline RNN-based methods. Our approach can be used for scaffold hopping, library expansion starting from a single molecule, and generating compounds with high predicted activity against biological targets.
Artificial Intelligence for Science in Quantum, Atomistic, and Continuum Systems
Jul 17, 2023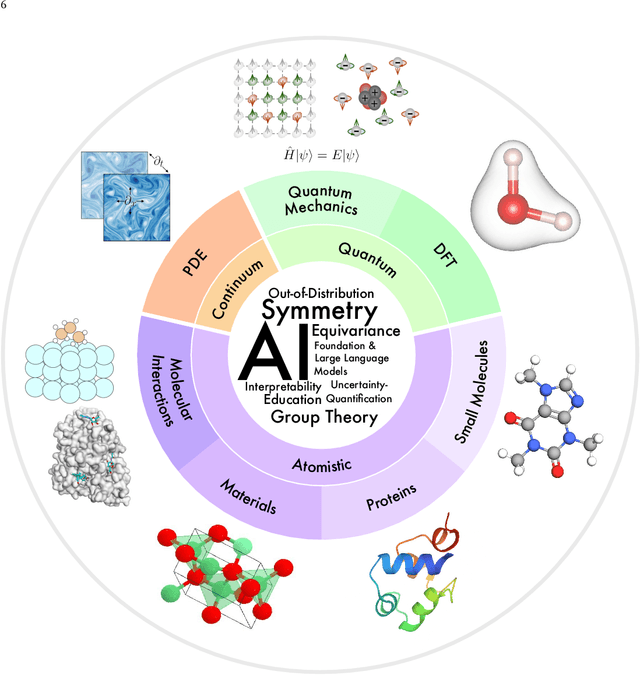

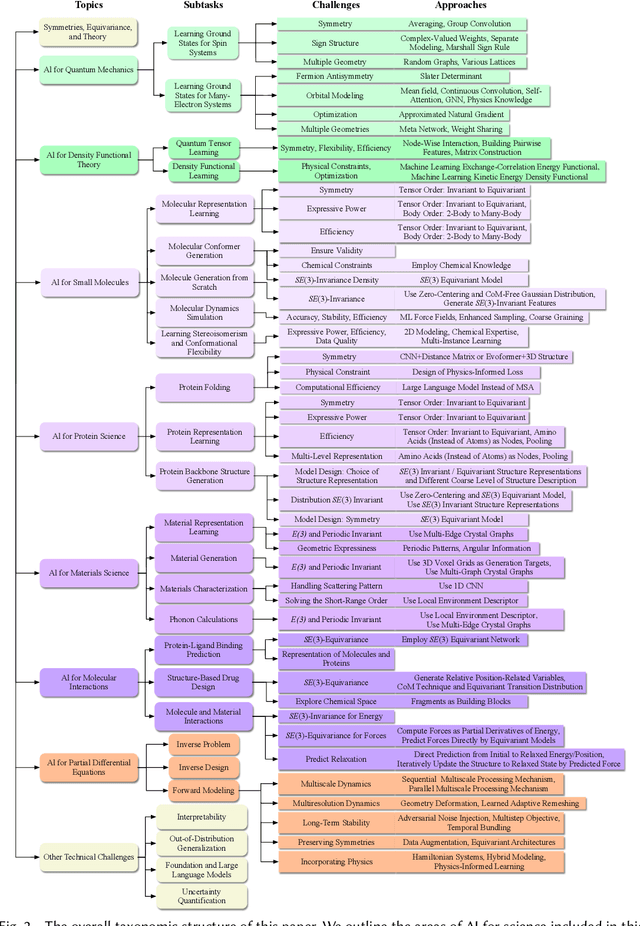
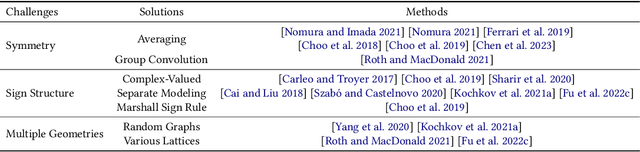
Advances in artificial intelligence (AI) are fueling a new paradigm of discoveries in natural sciences. Today, AI has started to advance natural sciences by improving, accelerating, and enabling our understanding of natural phenomena at a wide range of spatial and temporal scales, giving rise to a new area of research known as AI for science (AI4Science). Being an emerging research paradigm, AI4Science is unique in that it is an enormous and highly interdisciplinary area. Thus, a unified and technical treatment of this field is needed yet challenging. This paper aims to provide a technically thorough account of a subarea of AI4Science; namely, AI for quantum, atomistic, and continuum systems. These areas aim at understanding the physical world from the subatomic (wavefunctions and electron density), atomic (molecules, proteins, materials, and interactions), to macro (fluids, climate, and subsurface) scales and form an important subarea of AI4Science. A unique advantage of focusing on these areas is that they largely share a common set of challenges, thereby allowing a unified and foundational treatment. A key common challenge is how to capture physics first principles, especially symmetries, in natural systems by deep learning methods. We provide an in-depth yet intuitive account of techniques to achieve equivariance to symmetry transformations. We also discuss other common technical challenges, including explainability, out-of-distribution generalization, knowledge transfer with foundation and large language models, and uncertainty quantification. To facilitate learning and education, we provide categorized lists of resources that we found to be useful. We strive to be thorough and unified and hope this initial effort may trigger more community interests and efforts to further advance AI4Science.
PyTrial: A Comprehensive Platform for Artificial Intelligence for Drug Development
Jun 06, 2023



Drug development is a complex process that aims to test the efficacy and safety of candidate drugs in the human body for regulatory approval via clinical trials. Recently, machine learning has emerged as a vital tool for drug development, offering new opportunities to improve the efficiency and success rates of the process. To facilitate the research and development of artificial intelligence (AI) for drug development, we developed a Python package, namely PyTrial, that implements various clinical trial tasks supported by AI algorithms. To be specific, PyTrial implements 6 essential drug development tasks, including patient outcome prediction, trial site selection, trial outcome prediction, patient-trial matching, trial similarity search, and synthetic data generation. In PyTrial, all tasks are defined by four steps: load data, model definition, model training, and model evaluation, which can be done with a couple of lines of code. In addition, the modular API design allows practitioners to extend the framework to new algorithms and tasks easily. PyTrial is featured for a unified API, detailed documentation, and interactive examples with preprocessed benchmark data for all implemented algorithms. This package can be installed through Python Package Index (PyPI) and is publicly available at https://github.com/RyanWangZf/PyTrial.
Reinforced Genetic Algorithm for Structure-based Drug Design
Nov 28, 2022


Structure-based drug design (SBDD) aims to discover drug candidates by finding molecules (ligands) that bind tightly to a disease-related protein (targets), which is the primary approach to computer-aided drug discovery. Recently, applying deep generative models for three-dimensional (3D) molecular design conditioned on protein pockets to solve SBDD has attracted much attention, but their formulation as probabilistic modeling often leads to unsatisfactory optimization performance. On the other hand, traditional combinatorial optimization methods such as genetic algorithms (GA) have demonstrated state-of-the-art performance in various molecular optimization tasks. However, they do not utilize protein target structure to inform design steps but rely on a random-walk-like exploration, which leads to unstable performance and no knowledge transfer between different tasks despite the similar binding physics. To achieve a more stable and efficient SBDD, we propose Reinforced Genetic Algorithm (RGA) that uses neural models to prioritize the profitable design steps and suppress random-walk behavior. The neural models take the 3D structure of the targets and ligands as inputs and are pre-trained using native complex structures to utilize the knowledge of the shared binding physics from different targets and then fine-tuned during optimization. We conduct thorough empirical studies on optimizing binding affinity to various disease targets and show that RGA outperforms the baselines in terms of docking scores and is more robust to random initializations. The ablation study also indicates that the training on different targets helps improve performance by leveraging the shared underlying physics of the binding processes. The code is available at https://github.com/futianfan/reinforced-genetic-algorithm.
MolGenSurvey: A Systematic Survey in Machine Learning Models for Molecule Design
Mar 28, 2022



Molecule design is a fundamental problem in molecular science and has critical applications in a variety of areas, such as drug discovery, material science, etc. However, due to the large searching space, it is impossible for human experts to enumerate and test all molecules in wet-lab experiments. Recently, with the rapid development of machine learning methods, especially generative methods, molecule design has achieved great progress by leveraging machine learning models to generate candidate molecules. In this paper, we systematically review the most relevant work in machine learning models for molecule design. We start with a brief review of the mainstream molecule featurization and representation methods (including 1D string, 2D graph, and 3D geometry) and general generative methods (deep generative and combinatorial optimization methods). Then we summarize all the existing molecule design problems into several venues according to the problem setup, including input, output types and goals. Finally, we conclude with the open challenges and point out future opportunities of machine learning models for molecule design in real-world applications.
Differentiable Scaffolding Tree for Molecular Optimization
Sep 22, 2021



The structural design of functional molecules, also called molecular optimization, is an essential chemical science and engineering task with important applications, such as drug discovery. Deep generative models and combinatorial optimization methods achieve initial success but still struggle with directly modeling discrete chemical structures and often heavily rely on brute-force enumeration. The challenge comes from the discrete and non-differentiable nature of molecule structures. To address this, we propose differentiable scaffolding tree (DST) that utilizes a learned knowledge network to convert discrete chemical structures to locally differentiable ones. DST enables a gradient-based optimization on a chemical graph structure by back-propagating the derivatives from the target properties through a graph neural network (GNN). Our empirical studies show the gradient-based molecular optimizations are both effective and sample efficient. Furthermore, the learned graph parameters can also provide an explanation that helps domain experts understand the model output.