 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeXiaoning Qian
Complete and Efficient Graph Transformers for Crystal Material Property Prediction
Mar 18, 2024
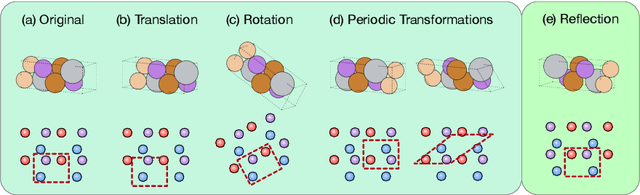
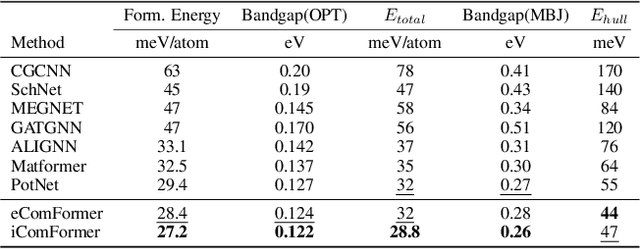
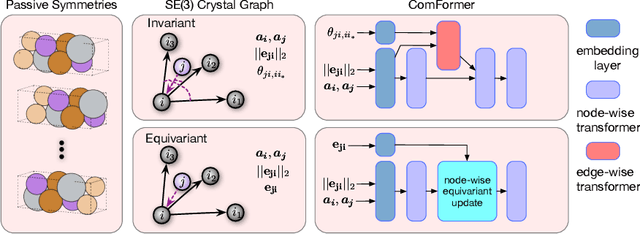
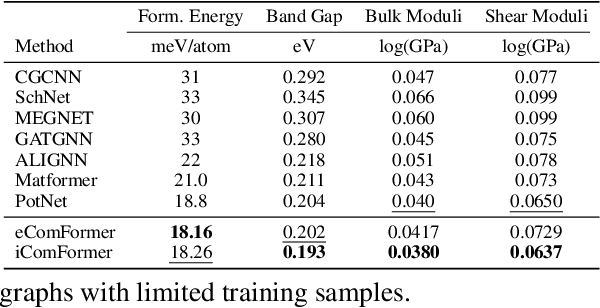
Crystal structures are characterized by atomic bases within a primitive unit cell that repeats along a regular lattice throughout 3D space. The periodic and infinite nature of crystals poses unique challenges for geometric graph representation learning. Specifically, constructing graphs that effectively capture the complete geometric information of crystals and handle chiral crystals remains an unsolved and challenging problem. In this paper, we introduce a novel approach that utilizes the periodic patterns of unit cells to establish the lattice-based representation for each atom, enabling efficient and expressive graph representations of crystals. Furthermore, we propose ComFormer, a SE(3) transformer designed specifically for crystalline materials. ComFormer includes two variants; namely, iComFormer that employs invariant geometric descriptors of Euclidean distances and angles, and eComFormer that utilizes equivariant vector representations. Experimental results demonstrate the state-of-the-art predictive accuracy of ComFormer variants on various tasks across three widely-used crystal benchmarks. Our code is publicly available as part of the AIRS library (https://github.com/divelab/AIRS).
Causal Bayesian Optimization via Exogenous Distribution Learning
Feb 06, 2024Maximizing a target variable as an operational objective in a structured causal model is an important problem. Existing Causal Bayesian Optimization (CBO) methods either rely on hard interventions that alter the causal structure to maximize the reward; or introduce action nodes to endogenous variables so that the data generation mechanisms are adjusted to achieve the objective. In this paper, a novel method is introduced to learn the distribution of exogenous variables, which is typically ignored or marginalized through expectation by existing methods. Exogenous distribution learning improves the approximation accuracy of structured causal models in a surrogate model that is usually trained with limited observational data. Moreover, the learned exogenous distribution extends existing CBO to general causal schemes beyond Additive Noise Models (ANM). The recovery of exogenous variables allows us to use a more flexible prior for noise or unobserved hidden variables. A new CBO method is developed by leveraging the learned exogenous distribution. Experiments on different datasets and applications show the benefits of our proposed method.
Dynamic Incremental Optimization for Best Subset Selection
Feb 06, 2024Best subset selection is considered the `gold standard' for many sparse learning problems. A variety of optimization techniques have been proposed to attack this non-smooth non-convex problem. In this paper, we investigate the dual forms of a family of $\ell_0$-regularized problems. An efficient primal-dual algorithm is developed based on the primal and dual problem structures. By leveraging the dual range estimation along with the incremental strategy, our algorithm potentially reduces redundant computation and improves the solutions of best subset selection. Theoretical analysis and experiments on synthetic and real-world datasets validate the efficiency and statistical properties of the proposed solutions.
Multi-modal Representation Learning for Cross-modal Prediction of Continuous Weather Patterns from Discrete Low-Dimensional Data
Jan 30, 2024World is looking for clean and renewable energy sources that do not pollute the environment, in an attempt to reduce greenhouse gas emissions that contribute to global warming. Wind energy has significant potential to not only reduce greenhouse emission, but also meet the ever increasing demand for energy. To enable the effective utilization of wind energy, addressing the following three challenges in wind data analysis is crucial. Firstly, improving data resolution in various climate conditions to ensure an ample supply of information for assessing potential energy resources. Secondly, implementing dimensionality reduction techniques for data collected from sensors/simulations to efficiently manage and store large datasets. Thirdly, extrapolating wind data from one spatial specification to another, particularly in cases where data acquisition may be impractical or costly. We propose a deep learning based approach to achieve multi-modal continuous resolution wind data prediction from discontinuous wind data, along with data dimensionality reduction.
Learning Active Subspaces for Effective and Scalable Uncertainty Quantification in Deep Neural Networks
Sep 06, 2023



Bayesian inference for neural networks, or Bayesian deep learning, has the potential to provide well-calibrated predictions with quantified uncertainty and robustness. However, the main hurdle for Bayesian deep learning is its computational complexity due to the high dimensionality of the parameter space. In this work, we propose a novel scheme that addresses this limitation by constructing a low-dimensional subspace of the neural network parameters-referred to as an active subspace-by identifying the parameter directions that have the most significant influence on the output of the neural network. We demonstrate that the significantly reduced active subspace enables effective and scalable Bayesian inference via either Monte Carlo (MC) sampling methods, otherwise computationally intractable, or variational inference. Empirically, our approach provides reliable predictions with robust uncertainty estimates for various regression tasks.
Artificial Intelligence for Science in Quantum, Atomistic, and Continuum Systems
Jul 17, 2023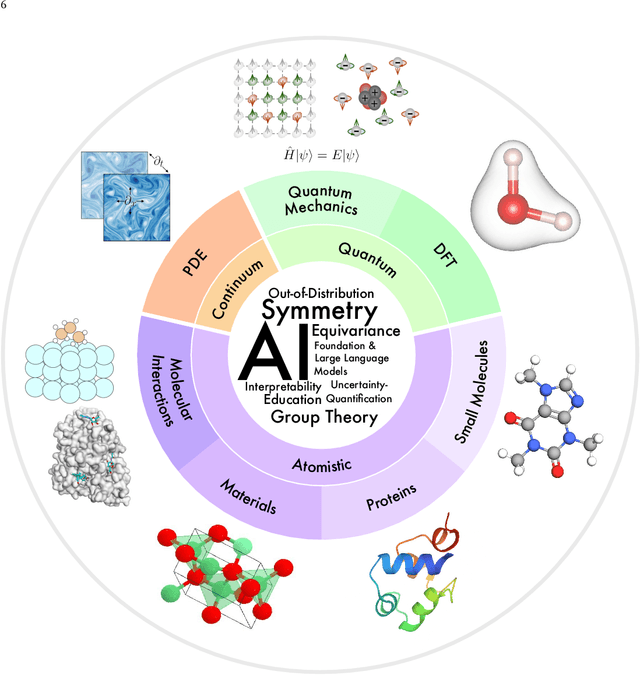

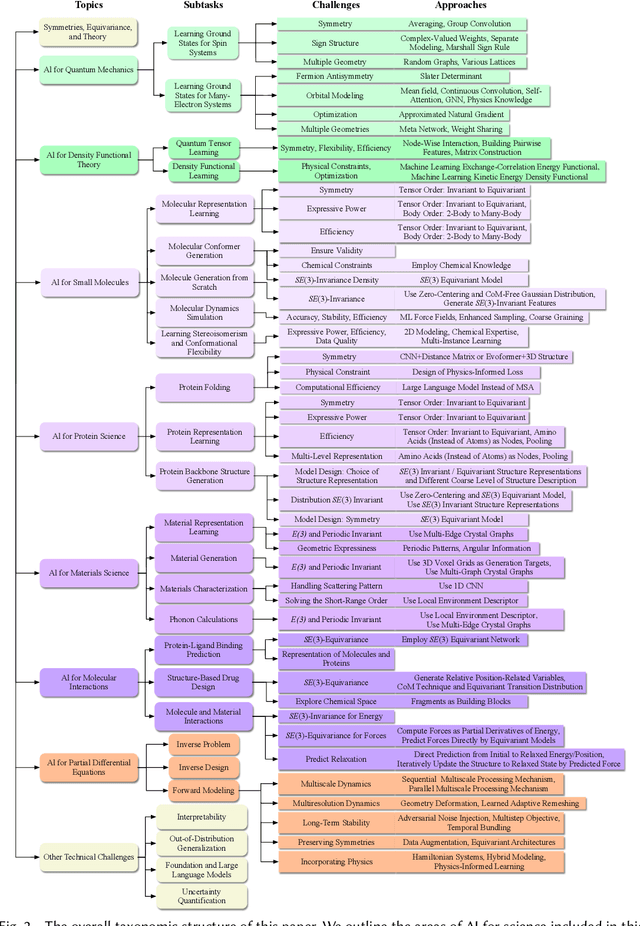
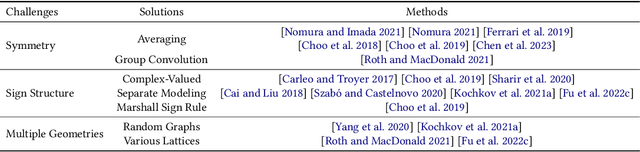
Advances in artificial intelligence (AI) are fueling a new paradigm of discoveries in natural sciences. Today, AI has started to advance natural sciences by improving, accelerating, and enabling our understanding of natural phenomena at a wide range of spatial and temporal scales, giving rise to a new area of research known as AI for science (AI4Science). Being an emerging research paradigm, AI4Science is unique in that it is an enormous and highly interdisciplinary area. Thus, a unified and technical treatment of this field is needed yet challenging. This paper aims to provide a technically thorough account of a subarea of AI4Science; namely, AI for quantum, atomistic, and continuum systems. These areas aim at understanding the physical world from the subatomic (wavefunctions and electron density), atomic (molecules, proteins, materials, and interactions), to macro (fluids, climate, and subsurface) scales and form an important subarea of AI4Science. A unique advantage of focusing on these areas is that they largely share a common set of challenges, thereby allowing a unified and foundational treatment. A key common challenge is how to capture physics first principles, especially symmetries, in natural systems by deep learning methods. We provide an in-depth yet intuitive account of techniques to achieve equivariance to symmetry transformations. We also discuss other common technical challenges, including explainability, out-of-distribution generalization, knowledge transfer with foundation and large language models, and uncertainty quantification. To facilitate learning and education, we provide categorized lists of resources that we found to be useful. We strive to be thorough and unified and hope this initial effort may trigger more community interests and efforts to further advance AI4Science.
Efficient Approximations of Complete Interatomic Potentials for Crystal Property Prediction
Jun 26, 2023



We study property prediction for crystal materials. A crystal structure consists of a minimal unit cell that is repeated infinitely in 3D space. How to accurately represent such repetitive structures in machine learning models remains unresolved. Current methods construct graphs by establishing edges only between nearby nodes, thereby failing to faithfully capture infinite repeating patterns and distant interatomic interactions. In this work, we propose several innovations to overcome these limitations. First, we propose to model physics-principled interatomic potentials directly instead of only using distances as in many existing methods. These potentials include the Coulomb potential, London dispersion potential, and Pauli repulsion potential. Second, we model the complete set of potentials among all atoms, instead of only between nearby atoms as in existing methods. This is enabled by our approximations of infinite potential summations with provable error bounds. We further develop efficient algorithms to compute the approximations. Finally, we propose to incorporate our computations of complete interatomic potentials into message passing neural networks for representation learning. We perform experiments on the JARVIS and Materials Project benchmarks for evaluation. Results show that the use of interatomic potentials and complete interatomic potentials leads to consistent performance improvements with reasonable computational costs. Our code is publicly available as part of the AIRS library (https://github.com/divelab/AIRS/tree/main/OpenMat/PotNet).
QH9: A Quantum Hamiltonian Prediction Benchmark for QM9 Molecules
Jun 15, 2023



Supervised machine learning approaches have been increasingly used in accelerating electronic structure prediction as surrogates of first-principle computational methods, such as density functional theory (DFT). While numerous quantum chemistry datasets focus on chemical properties and atomic forces, the ability to achieve accurate and efficient prediction of the Hamiltonian matrix is highly desired, as it is the most important and fundamental physical quantity that determines the quantum states of physical systems and chemical properties. In this work, we generate a new Quantum Hamiltonian dataset, named as QH9, to provide precise Hamiltonian matrices for 2,399 molecular dynamics trajectories and 130,831 stable molecular geometries, based on the QM9 dataset. By designing benchmark tasks with various molecules, we show that current machine learning models have the capacity to predict Hamiltonian matrices for arbitrary molecules. Both the QH9 dataset and the baseline models are provided to the community through an open-source benchmark, which can be highly valuable for developing machine learning methods and accelerating molecular and materials design for scientific and technological applications. Our benchmark is publicly available at https://github.com/divelab/AIRS/tree/main/OpenDFT/QHBench.
Efficient and Equivariant Graph Networks for Predicting Quantum Hamiltonian
Jun 08, 2023



We consider the prediction of the Hamiltonian matrix, which finds use in quantum chemistry and condensed matter physics. Efficiency and equivariance are two important, but conflicting factors. In this work, we propose a SE(3)-equivariant network, named QHNet, that achieves efficiency and equivariance. Our key advance lies at the innovative design of QHNet architecture, which not only obeys the underlying symmetries, but also enables the reduction of number of tensor products by 92\%. In addition, QHNet prevents the exponential growth of channel dimension when more atom types are involved. We perform experiments on MD17 datasets, including four molecular systems. Experimental results show that our QHNet can achieve comparable performance to the state of the art methods at a significantly faster speed. Besides, our QHNet consumes 50\% less memory due to its streamlined architecture. Our code is publicly available as part of the AIRS library (\url{https://github.com/divelab/AIRS}).
A Latent Diffusion Model for Protein Structure Generation
May 06, 2023



Proteins are complex biomolecules that perform a variety of crucial functions within living organisms. Designing and generating novel proteins can pave the way for many future synthetic biology applications, including drug discovery. However, it remains a challenging computational task due to the large modeling space of protein structures. In this study, we propose a latent diffusion model that can reduce the complexity of protein modeling while flexibly capturing the distribution of natural protein structures in a condensed latent space. Specifically, we propose an equivariant protein autoencoder that embeds proteins into a latent space and then uses an equivariant diffusion model to learn the distribution of the latent protein representations. Experimental results demonstrate that our method can effectively generate novel protein backbone structures with high designability and efficiency.